Abstract
The COVID-19 pandemic has paralleled the great Spanish flu pandemic of 1918–1919 in the United States. Previous historical accounts have strongly suggested a post-viral syndrome and, currently, a post-COVID-19 viral syndrome is unquestionable, which shares many of the characteristics of myalgic encephalomyelitis/chronic fatigue syndrome that is present globally. The original term for this post-acute sequela of SARS-CoV-2 (PASC) was termed long haulers by those who were affected with this syndrome and it is now termed long COVID (LC) or PASC. International researchers and clinicians are desperately trying to better understand the pathobiological mechanisms possibly involved in this syndrome. This review aims to summarize many of the cumulated findings associated with LC/PASC and provides supportive and representative illustrations and transmission electron micrographic remodeling changes within brain tissues associated with a stress type of injury as occurs in the classic db/db and novel BTBR ob/ob obesity and diabetes mellitus mice models. These models are utilized to merely provide a response to metabolic stress injury wound healing mechanisms that are also present in humans. This review posits that neuroglial activation and chronic neuroinflammation may be a common denominator for the development of the complex LC/PASC syndrome following acute COVID-19 due to SARS-CoV-2.
Keywords:
astrocyte; BBB; BCSFB; brain endothelial cell; chemokines; COVID-19; cytokines; long COVID; neuroglia; PASC; ultrastructure 1. Introduction
Fortunately, large numbers of individuals have now recovered from the acute coronavirus disease-19 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Unfortunately, there are growing numbers and concerns regarding a subset of these recovered individuals who have varied, persistent, and reoccurring symptoms relating to this pandemic that have termed themselves as ‘long haulers’, while the medical community has now termed this condition as long-haul COVID—long COVID (LC) or post-acute sequela of SARS-CoV-2 (PASC) [1,2]. Hereafter, the terms LC and PASC will be referred to as LC/PASC. It is important to note at this time that there is still not a standardized definition of LC/PASC; however, in the broadest sense, these individuals represent a failure to return to a baseline state of health after recovering from acute COVID-19 and encompass various physical, neurologic, and neuropsychiatric symptoms lasting from weeks to months following the acute infection of COVID-19. Typical symptoms of LC/PASC include fatigue (central/cognitive, or mental and peripheral/muscular or physical), shortness of breath, chest pain, “brain fog” (impaired cognition with difficulty in maintaining focus), sleep disorders (insomnia), fevers, gastrointestinal symptoms (diarrhea, nausea and/or vomiting), anxiety, and depression that can persist from weeks to months and can range from mild to incapacitating (Figure 1) [1,2].
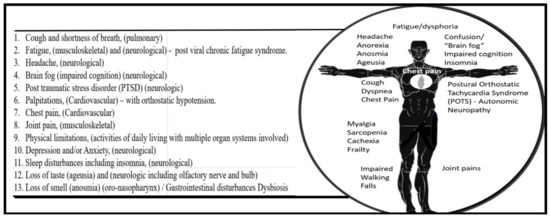
Figure 1.
The above listing represents at least 13 of most common associated multiorgan symptoms of individuals following the normal amount of time for healing of this disease known as long COVID or post-acute sequelae of SARS-CoV-2 (LC/PASC). In some cases, such as gastrointestinal disturbances and microbiome dysbiosis, there may be an interaction via a gut–brain axis, which may result in neurological/neuropsychiatric symptoms. Importantly, this list affects individuals of varying ages and occurs in some individuals in reoccurring waves and not a continuous on-going fashion. Importantly, note that at least 7 of the 13 most common listed symptoms involve neurological systems and mechanisms. In some cases, even new symptoms arise well after the time of infection or evolve over time that are capable of interfering with normal activities of daily living [3]. The standing man depicts the various physical locations where some of these LC/PASC clinical symptoms occur in humans and note that at least eight of these symptoms may relate to the central nervous system.
The NIH director, Francis Collins, has recently reported on the findings from a NIH workshop entitled “Workshop on Post-Acute Sequelae of SARS-CoV-2 (PASC)”, in which he summarized what is known about post-COVID-19 symptoms and identified key gaps in the knowledge surrounding this [3]. In addition to this workshop, it was announced previously in December 2020 that the United States Congress has provided 1.15 billion dollars in funding to be made available over four years for the NIH to support research into the prolonged health consequences of SARS-CoV-2 infection. Some of the key questions in regards to this funding include: (1) “What does the spectrum of recovery from SARS-CoV-2 infection look like across the population?”; (2) “How many people continue to have symptoms of COVID-19, or even develop new symptoms, after acute SARS-CoV-2 infection?”; (3) “How many people continue to have symptoms of COVID-19, or even develop new symptoms, after acute SARS-CoV-2 infection?”; (4) “What is the underlying biological cause of these prolonged symptoms?”; (5) “What makes some people vulnerable to this but not others?”; and (6) “Does SARS-CoV-2 infection trigger changes in the body that increase the risk of other conditions, such as chronic heart or brain disorders?” [3]. It is anticipated that these questions might stimulate further research to investigate opportunities that will focus on clinical trials to test treatment strategies regarding PASC symptoms and, thus, promote recovery [3]. Additionally, there exists the need to access the emotional, behavioral, and societal impact of the COVID-19 pandemic across all nations [4].
Specific time frames of LC/PASC have not been universally defined at the time of this manuscript’s preparation; however, there seems to be an emerging consensus of greater than four weeks post-acute COVID-19 [5]. However, authors speculate that the time frame may ultimately be between four to eight weeks and that this time frame might be more in line with the normal healing time as occurs in the response to injury wound healing mechanisms that are at play in response to non-central nervous system tissue/organ injuries [6,7,8,9,10,11]. The central nervous system (CNS) is unique in its response to injury wound healing mechanisms since the brain does not have fibrocytes/fibroblasts as in peripheral tissues, which is important for fibrosis and scar formation; however, the brain possesses neuroglial astrocyte(s) (ACs) that are capable of undergoing activation and reactive astrogliosis that is responsible for the protective glial scar formation in the brain following injury (Figure 2) [10,11].
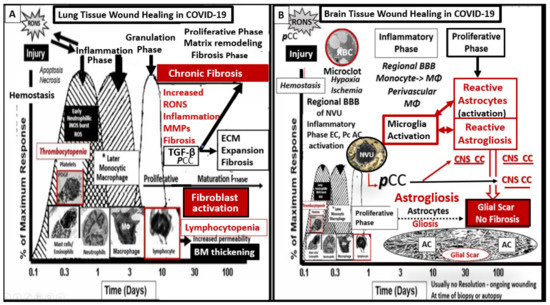
Figure 2.
Response to injury wound healing mechanisms in the brain compared to peripheral tissues. Panel A illustrates the timeline of response to injury wound healing mechanisms in peripheral tissues, such as in the skin or the lungs, and includes the various cells and mechanisms involved, which progress to fibrosis. Panel B depicts the divergence in the wound healing response to injury in the central nervous system (CNS). Note that the injury in this case is the microclot/thrombosis and/or microhemorrhage (not shown) of regional brain capillary neurovascular unit(s) (NVUs) in addition to the excessive peripheral proinflammatory cytokines and chemokines (pCC) from the cytokine, viral/virion, redox, and the inflammatory thromboembolic storms as in COVID-19 injury. In the obese diabetic db/db and ob/ob models, the CNS injury may also be due to glucotoxicity and excessive reactive oxygen and nitrogen species (RONS). The resident immune cell in the CNS is the neuroglial microglia cell (MGC). MGCs support the initial acute and chronic inflammatory responses to injury. However, the peripheral monocyte/monocyte-derived macrophages (MΦ) from the regionally activated NVUs are supportive to the resident MGCs to clear debris from CNS microthrombus/clot or micohemorrhage—stroke ischemic and hypoxic injury such that the peripheral inflammatory systems aid the resident MGC population in the CNS inflammatory response to injury. Notably, the NVUs-activated endothelial cells (ECs), pericytes (Pcs), and astrocytes (ACs) of the blood–brain barrier (BBB) may contribute to the CNS cytokines/chemokine (CNSCC) activation as well as the choroid plexus, which forms the blood cerebrospinal fluid barrier (BCSFB) and the meningeal arachnoid BCSFB. Importantly, note that the CNS tissues do not have fibrocytes/fibroblasts in the CNS parenchymal tissues and, therefore, do not form a fibrotic scar as in peripheral tissues such as the lung or skin in Panel A. Instead, the CNS utilizes the glial astrocytes (ACs) to undergo the process of astrogliosis in order to form a glial scar to protect the adjacent neuropil from the injury and damage caused by a micro thrombotic/micro hemorrhagic stroke. Unfortunately, the activated CNS glial cells add to the CNSCC (IL-6, IL-1β, TNFα, growth factor TGF-β) (IL-10 anti-inflammatory)/chemokine (CXCL2 -MCP-1 + others) toxic inflammatory damage to the CNS. Within the glial scar core, the extracellular matrix (ECM) remodeling over time helps to fill in the void created by ischemic injury to cells and the MGCs and macrophage (MΦ) phagocytosis. Additionally, activated glial cells contribute to the ongoing CNS proinflammatory damage to the CNS neurons and neuronal synapses, which may result in impaired synaptic transmission and impaired cognition in long COVID and may support many of the neurological symptoms listed in Figure 1. Panel A adapted with permission CC by 4.0 [9]. AC = astrocyte; BBB = blood–brain barrier; BM = basement membrane; CNS = central nervous system; CNSCC = central nervous system cytokines/chemokines; CXCL2/MCP-1 = Chemokine (C-X-C motif) ligand 2/ monocyte chemotactic protein-1; EC = endothelial cell; ECM = extracellular matrix; IL-1β = interleukin-1 beta; IL-6 = interleukin-6; IL-10 = interleukin-10 (anti-inflammatory cytokine); MΦ = macrophage; NVU = neurovascular unit; pCC= peripheral pro-inflammatory cytokines/chemokines; Pc = pericyte; RONS = reactive oxygen nitrogen species; ROS =reactive oxygen species; TGF-β = transforming growth factor-beta; TNF-α = tumor necrosis factor alpha.
Some feel that the healing time for CNS injuries may take longer than non-CNS injuries, such as in the skin and pulmonary tissues [10]. Importantly, it has been determined that there was not replication-competent SARS-CoV-2 isolated after three weeks in acute COVID-19 infections [12,13].
Peripheral inflammation of the immune–brain axis is thought to be bidirectional in humans in regards to immune system function. COVID-19 has been shown to have a dysregulated immune system response to SARS-CoV-2 and has highlighted the important role of cytokines and chemokines since it is associated with a profound hypercytokinemia that has been termed cytokine storm [14]. Cytokines may be defined as a broad and loose category of small immunomodulatory peptides, proteins, or glycoproteins (~5–30 kDa) which are important in cell signaling including growth, maturation, and responsiveness of particular cell populations, and chemokines are cytokines with chemotactic properties that induce leukocyte migration and transmigration across brain endothelial cells (BECs) in the CNS [15].
This review will focus on the peripheral pro-inflammatory cytokines/chemokines (PCC) and the central nervous system (CNS) proinflammatory cytokines/chemokines (CNSCC) and their effects on the neurovascular unit (NVU) blood–brain barrier (BBB) and the choroid plexus (CP) blood–cerebrospinal barrier (BCSFB), neuroglial cells (microglia cell(s) (MGCs), astrocytes (ACs), and oligodendrocytes (OLGs)); the response to injury wound healing mechanisms; and LC/PASC chronic neuroinflammation. In addition to the acute COVID-19 inflammatory thromboembolic state, the effect of chronic inflammation in CNS and its associated microthrombi/microbleeds, which result in decreased cerebral blood flow (CBF) and ischemia and ischemia/reperfusion injury to evoke even further neuroinflammation to result in LC/PASC, will be discussed. Furthermore, the importance of impaired folate-mediated one-carbon metabolism will be discussed and related to the LC/PASC post-COVID-19 syndrome in addition to the possibility of hypoadrenalism.
There is a paucity of COVID-19 autopsied brain tissue that have been properly prepared to study ultrastructure changes. However, the changes seen among neuroinflammatory conditions likely have a number of similarities. One of the best studied conditions from the ultrastructural point of view that has a neuroinflammatory component are the models of type 2 diabetes (T2DM). Therefore, transmission electron microscopy (TEM) works on models of T2DM (db/db and BTBR ob/ob) which are utilized to illustrate stress-induced ultrastructural remodeling. These TEM images are utilized in order to be merely representitive of the possible similar stress injury-induced remodeling changes that may also be present in LC/PASC as a result of previous COVID-19 infections with neuroglial microglia, astrocyte activation, and neuroinflammation.
2. Peripheral—Pro-Inflammatory Cytokines/Chemokines (PCC), Brain Barriers and CNS Cytokines/Chemokines (CNSCC): Mechanisms of Chronic Neuroinflammation and Impaired Cognition in LC/PASC
Because SARS-CoV-2 binds to the cellular angiotensin converting enzyme 2 (ACE2) receptor that is present in multiple organs systems, LC/PASC has a multifactorial pathogenesis which incorporates chronic inflammation both in the peripheral organ systems as well as in the CNS [16]. Neuronal system dysfunction, specifically including the activation of neuroglial cells (microglia and astrocytes), is associated with neuroinflammation and impaired cognition, and the activation/dysfunction of brain endothelial cells are associated with subsequent inflammatory thromboembolism. Thus, the activation of neuroglial microglia and astrocytes are thought to be key pathogenetic mechanisms in LC/PASC [16].
The PCC’s are thought to be elevated in LC/PASC individuals and they may occur in a continuous process or in recurrent wave-like processes that affect the brain (Figure 3) [16,17].

Figure 3.
Possible sequence of neurologic sequelae in LC/PASC due to activation of peripheral cytokine/chemokines (pCC) and Central Nervous System Cytokine/Chemokines (CNSCC). BECs = brain endothelial cells; ECs = endothelial cells; NALT = nasopharynx- associated lymphoid tissue; MALT = mucosa-associated lymphoid tissue. Pcs = pericytes.
Phetsouphanh et al. [17] have presented findings from the adaptation of evidence-informed complex population health interventions for implementation and/or re-evaluation in new contexts (ADAPT) study that demonstrated a uniquely prolonged effect on aspects of both the innate and adaptive immune systems. They further suggested that these findings may be driving the LC/PASC syndrome. They found elevated type I interferons and type III interferon levels at eight months in LC/PASC and suggested that these findings demonstrated ongoing inflammation in contrast to those without LC/PASC [17]. Additionally, they studied a prospective cohort of individuals with LC/PASC (the ADAPT study) and compared results to age/gender-matched subjects without LC/PASC, healthy donors, and individuals infected with other non-SARS CoV2 human coronaviruses (the ADAPT-C study) and found an elevated diffuse serum inflammatory cytokine profile in symptomatic long COVID subjects that was maintained at eight months post-infection, which was not observed in asymptomatic COVID-19 survivors. This elevated cytokine profile in LC/PASC individuals consisted of an elevation of type I and III interferon levels, which are considered to be antiviral cytokines [17]. Interestingly, type I interferons (IFN) consist of IFN-α, IFN-β, IFN-ε, IFN-κ, and IFN-ω that, in humans, bind to the ubiquitously expressed type I IFN receptor (IFNAR), and type III interferons consist of IFN-λ1 (IL-29), IFN-λ2 (IL-28a), and IFN-λ3 that bind to the type III receptor (IFNLR) that are preferentially expressed on epithelial and certain myeloid cells [18].
Additionally, Low et al. have examined the role of PCC and binding to the brain endothelial cell receptors with subsequent activation of the blood–brain barrier, brain endothelial cells, pericytes, and astrocytes in producing and contributing to increased CNSCC, which result in chronic neuroinflammation as well as the additional generation of reactive oxygen/nitrogen species (RONS) [19]. Importantly, oxidative stress and RONS induce neuroinflammation via activation of nuclear factor kappa B (NF-κB) with the creation of a RONS–vicious inflammatory cycle since neuroinflammation begets RONS in the central nervous system and RONS begets neuroinflammation [20]. Furthermore, during the earlier acute COVID-19 phase the central nervous system microglia cells may be already primed and capable of elaborating even more cytokines and fueling this vicious cycle between RONS and neuroinflammation with increased CNSCC. These mechanisms may result in even more chronic ongoing neuroinflammation and result in central nervous system-mediated LC/PASC with central nervous system symptoms of fatigue, malaise, fever, impaired cognition, memory loss, dysphoria, and depression associated with LC/PASC [21]. While papers by Phetsouphanh et al. and Low et al. are currently in preprint format, they are each strong papers and are highly referenced [17,19].
2.1. Peripheral Proinflammatory Cytokine/Chemokine (PCC) Effects on the Neurovascular Unit (NVU): Activation of Neuroglia (Microglial, Astrocytes and Oligodendrocytes)
As the peripheral systemic PCC perfuse the cerebral microcirculation, they will attach to their respective specific receptors on brain endothelial cell(s) (BECs) and, in turn, will activate BECs, pericytes, and astrocytes that comprise the NVU blood–brain barrier interface. In turn, these NVU cells will release central nervous system-derived cytokines/chemokines (CNSCC) resulting in the activation of the neuroglia, consisting of the central nervous system microglial cells, astrocytes, and oligodendrocytes, which will also add to the overall burden of increased CNSCC and neuroinflammation [21]. Further, Erickson et al. [22] have demonstrated the transport of the chemokine CCL11 across the BBB, and Quaranta et al. have recently presented a poster entitled: Interactions of Chemokines with the Blood–Brain Barrier at the American Society for Pharmacology and Experimental Therapeutics (ASPET) conference, which demonstrated that the CCL2 and CCL5 can cross the BBB by a non-saturable transport system (personal communication with Daniel V. Quaranta of the Banks and Erickson laboratory).
PCC are known to modulate the inflammatory immune response throughout the body and a dysregulation or excessive production that occurs in the acute COVID-19 cytokine storm along with the residual PCC are known to be a central feature for the development of central nervous system neuroinflammation, neurodegeneration, and dysmyelination including myelin remodeling in the central nervous system via activation of the neuroglia and their addition to the CNSCC pool (Figure 3) [23].
2.2. Microglia Cells (MGCs)
Microglia cells (MGCs) are the mesodermal yolk-sac-derived resident innate immune cells of the brain and possess a unique quality to undergo rapid changes in their diversified spectrum of varying ultrastructural phenotypes. They may exist in a ramified state, a polarized activated amoeboid state, and an aged senescent morphological state [24]. Ramified microglia cell(s) (rMGCs) (M-2 polarization-like state) serve continuously as the housekeepers and cleaners of normally accumulating cellular and metabolic debris. They are the innate immune cells of the central nervous system and are in constant surveillance of their surroundings in order to detect any type of damage or injury within the central nervous system including the neurovascular unit and the blood–brain barrier (Figure 4 and Figure 5A) [24].
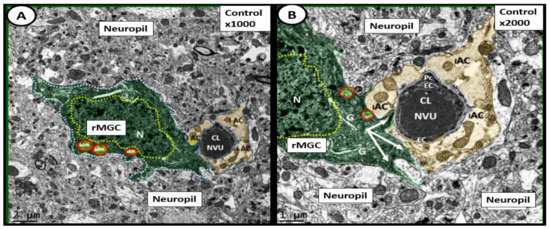
Figure 4.
Ramified surveilling microglia cells (rMGCs) in control (non-diabetic) C57BL/6J mice at 20-weeks of age. This image depicts the normal ultrastructure morphology of the ramified surveilling microglia cell (rMGC) and its relationship to the neurovascular unit (NVU) and neuropil. Panel A depicts the morphologic phenotype of a rMGC (pseudo-colored green) that is interrogating and surveilling the capillary NVU in cortical layer III. Panel B is a higher magnification of Panel A and one can readily note how the cytoplasmic processes of the rMGC have inserted beneath and abutted the supportive intact astrocytes (iAC) (pseudo-colored golden) in order to detect any danger or damage signals and clean any accumulating debris. rMGCs are known to have a stippled outer chromatin electron density and also a diffuse inner stippled chromatin arrangement of their nuclei. Note the aberrant mitochondria (aMt) numbering three in Panel A and two in Panel B (pseudo-colored yellow with red outline), which are in contrast to the multiple aMt in the diabetic db/db models. Magnification ×1000; scale bar = 2 μm in Panel A and magnification ×2000; scale bar = 1 μm in Panel B. Figures adapted with permission by CC 4.0 [22]. CL = capillary lumen; EC = endothelial cell; G = Golgi body; iAC= intact astrocyte; N= nucleus: Pc= pericyte.
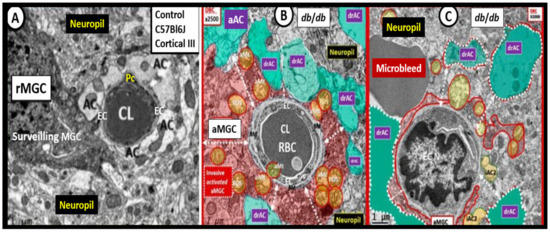
Figure 5.
Activated microglia cells (aMGCs) and astrocytes (ACs) in female diabetic db/db mice at 20 weeks of age. Panel A illustrates the ramified MGC (rMGC) that is surveilling the neurovascular unit blood–brain barrier (NVU BBB) interface in cortical layer III of a control C57BL/6J mouse at 20 weeks of age. Panel B demonstrates an activated MGC (aMGC), which appears to be totally encompassing the NVU BBB and note the swollen aberrant mitochondria (aMt) (pseudo-colored yellow responsible for mitochondria-derived increased reactive oxygen nitrogen species (RONS). Furthermore, note the detached and retracted activated astrocytes (drACs) (pseudo-colored cyan). Panel C also depicts drACs (pseudo-colored cyan) and note the partial encroachment of an aMGC. This capillary lumen appears nearly closed and also depicts a nearby well-demarcated grey homogeneous region compatible with a microbleed. Panels A–C are from cortical layer III in the frontal cortex just anterior to the midline of the brain in the diabetic db/db mouse at 20 weeks of age. Importantly, the tight and adherens junctions (TJ/AJ) are attenuated and/or lost in the female db/db mouse models at 20 weeks (not shown). Magnification ×2000; scale bar = 1 μm Panels A–C. Figures adapted with permission by CC 4.0 [24].
Ramified microglia cells (rMGCs) stand ‘on guard’ and are ready at a moment’s notice to morphologically shape shift into a M-1 polarization amoeboid-activated microglia (aMGCs) (Figure 5B,C), wherein their elongated cytoplasmic extensions become shorter, thicker, and more mobile. Additionally, there are changes within the aMGCs mitochondria in that they become increased in number, swollen, lose their mitochondria electron density within the matrix and fragment smudge, or lose their cristae. Normally microglia cells are in a ramified cleaning state and are in constant surveillance of their CNS surroundings including the neurovascular unit blood–brain barrier interface (Figure 4); however, when they are activated via PCCs/CNSCCs they will become activated to an M-1-like phenotype—aberrant MGCs (aMGCs) (Figure 5B,C) and become capable of generating reactive oxygen/nitrogen species, proteases, and will also add to the pool of increased secreted CNSCC [24]. Importantly, the aMGCs are responsible for the rearrangement attenuation and/or loss of the tight and adherens junctions (TJ/AJ) of the neurovascular unit blood–brain barriers of brain endothelial cells. This perturbation of the brain endothelial cells allows for increased adherence and transcellular and paracellular transmigration of innate immune neutrophils and monocytes (monocyte-derived macrophages with the brain) and adaptive immune lymphocytic cells into the central nervous system [24,25]. Interestingly, rMGCs can be primed by any central nervous system inflammatory insult and, upon being rechallenged, these primed MGCs are capable of becoming hyperactive aMGCs (capable of undergoing even further spectral changes) that produce even more toxic CNSCC and reactive oxygen nitrogen species than when they were initially activated the first time and, thus, implies a two-hit hypothesis [24,26]. This two-hit hypothesis may be helpful to explain why some COVID-19 patients, such as those with obesity and T2DM, are at greater risks for the development of more severe complications related to COVID-19, i.e., their microglia cells may be already primed from exposure to a chronic low-grade inflammation arising from adipose tissue depots, and COVID-19 will act as a second hit via an increase in PCC. The microglial cell two-hit hypothesis responsible for the hyperactive/exaggerated response of previously sensitized or primed aMGCs may also be in play in those with LC/PASC and help to explain some of their delayed neurological symptoms [26].
2.3. Astrocytes (ACs)
Astrocytes (ACs) are the most abundant cell in the human brain and with their vast connections to multiple other cell types including themselves are the master connecting and communicating cells within the human brain (Figure 6) [25,27,28,29]. This large AC cellular presence in the brain with their vast cell–cell communications via gap junctions may be viewed as the brain’s functional syncytium [25,30].
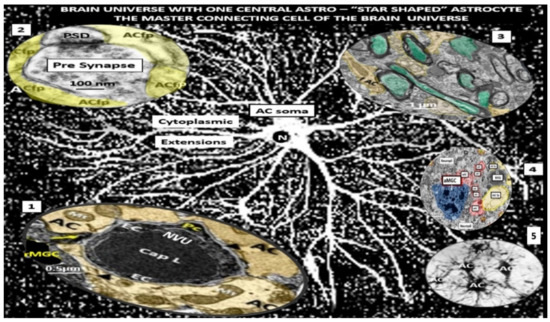
Figure 6.
Astrocyte (AC) connections to other cells within the brain universe. This montage illustration of hand-drawn images and transmission electron micrographs allows one to observe the resident non-activated AC soma as seen in the center of this image with multiple cytoplasmic extensions that connect to virtually every cell within the brain universe. This may, thus, be considered the master connecting cell of the central nervous system (CNS) against a background of distant white dots representing even more ACs stars within the brain universe from control C57BL/6J models (hand-drawn computer-assisted illustration from toluidine blue stained images). Importantly the AC-to-AC connections via gap junctions allow them to form a syncytium to coordinate brain function and proper cognition. Insert 1 depicts the protoplasmic AC foot processes (pseudo-colored golden) connecting to the neurovascular unit (NVU) capillary, which allows for neurovascular coupling in cortical layer III in a non-diabetic control C57BL/6J mouse model. Insert 2 illustrates the protoplasmic AC foot processes cradling a pre- and post-synaptic neuron and its importance to the tripartite cradle synapse in cortical layer III in a control C57BL/6J model. Insert 3 demonstrates the connection of the fibrous AC (pseudo-colored yellow-gold) to myelinated axons with axoplasm (pseudo-colored cyan) of neurons in white matter in a control C57BL/6J. Insert 4 illustrates the lost connections between an activated microglial cell (aMGC) (pseudo-colored blue) and multiple activated detached ACs (pseudo-colored red) adjacent to a neurovascular unit (NVU) with intact non-activated ACs (pseudo-colored yellow) in diabetic db/db model cortical layer III at 20 weeks of age. Insert 5 demonstrates AC-to-AC connections in cortical layer III in control models (hand-drawn computer-assisted illustration of light microscopic toluidine blue stained images from control C57BL/6J models). Only Inserts 1–4 have scale bars of 0.5 μm, 100 nm, 1 μm, and 2 μm, respectively. The background and insert 5 are hand-drawn computer-assisted images derived from control C57BL/6J models and, therefore, do not have a scale bar. Inserts are adapted cropped images with permission by CC 4.0 [23,24,25]. ACfp—AC = protoplasmic astrocyte foot processes; Cap = capillary; fAC = fibrous astrocyte foot processes; N = nucleus; PSD = post-synaptic density.
Astrocytes perform multiple important functions in the brain to provide maintenance of extracellular homeostasis, including glutamate uptake and recycling; potassium buffering; supply of energy substrates, especially the astrocyte neuron coupling involving the astrocyte neuron lactate shuttle (ANLS) hypothesis; spatiotemporal neurovascular unit coupling between neuronal activity; and cerebral blood flow, providing functional hyperemia (Figure 5B,C and Figure 6), pH buffering, and antioxidant defense against oxidative stress via glutathione (GSH); and recycling of the micronutrient ascorbate, to name a few [31].
Peripheral immune activation, such as PCC, can instigate rapid and concerted responses of CNSCC neuroglia regulated through fine-tuned communications between astrocytes, microglia, and mural cells of brain barriers including brain endothelial cells and pericytes. This also propagates inflammatory signals over the brain, and induces physiological and, importantly, behavioral changes that may correspond to sickness behavior or more severe impaired cognition characteristic of the LC/PASC, as noted in more severe sepsis associated encephalopathy models [32]. One remodeling characteristic of astrocyte activation in the diabetic db/db model was detachment and retraction from the basement membrane of the neurovascular unit, which could result in the uncoupling of the neurovascular unit and impair regional vasodilation when regional neurons are active and, thus, impair regional cerebral blood-flow-producing regional hypoxia (Figure 5B,C) [25]. Reactive astrocytes following injury are initially protective; however, as the response to injury progresses, the astrocytes will remodel to become a more astrogliopathic phenotype with retraction of their cytoplasmic extensions and lose their communication with not only the neurovascular units but also neurons, synapses, and their own cell–cell connections [25,32].
2.4. Oligodendrocytes (OLGs) and Myelin
Oligodendrocytes (OLGs) play an important role in myelin synthesis, wrapping, and compaction of myelinated axons and are remodeled when there is injury by T2DM with excess of reactive oxygen/nitrogen species and CNSCC [33]. Further, activation of microglia and astrocytes will also have effects on the function of the OLGs. Previously, our lab has found prominent remodeling of OLGs with increased nuclear chromatin condensation and volume with increased numbers of active myelination sites of the cytoplasm in transition zones. Furthermore, marked aberrant myelination was noted with outer myelin lamellae sheath with splitting, separation, ballooning, and aberrant mitochondria in grey matter cortical layer III and similar myelin remodeling changes with marked disarray with additional axonal collapse in transitional zones between the grey and white matter in the diabetic db/db as compared to the non-diabetic controls at 20-weeks of age (Figure 7) [33].
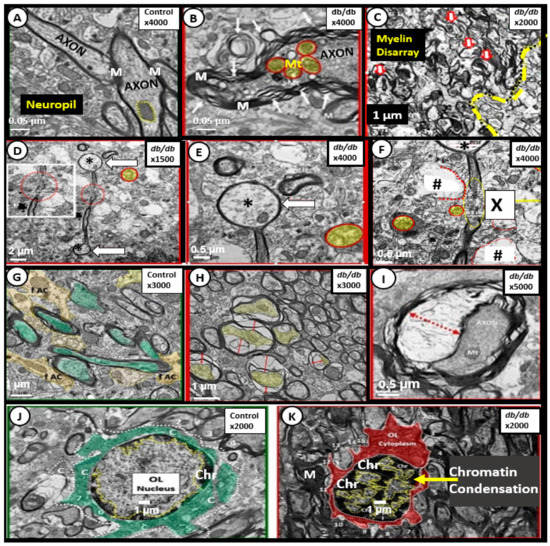
Figure 7.
Aberrant oligodendrocyte and myelin remodeling in the obese insulin resistant diabetic 20-week-old mice. Panel A illustrates the appearance of normal myelin ensheathing axons in the cortical grey matter of a control non-diabetic C57BL/6J model. Panels B and C depict marked splitting and separation of myelin and marked disarray of myelin in cortical layers and the transition zone between the grey and white matter, respectively, in the diabetic db/db models. Note the yellow outlined in red smudged aberrant mitochondria (Mt). Panels D–F depict the terminal ballooning (arrows) of myelinated axons (asterisks). Note the detached and retracted supportive ACs in Panel F (hashtags) and a region of total axon and myelin disruption (X). Panels E and F are higher magnifications of Panel D. Panel G illustrates the normal myelinated axons (pseudo-colored green) with tightly abutting supportive astrocyte(s) (ACs) in control non-diabetic C57BL/6J models. Panels H and I depict axonal collapse (yellow in Panel H) with red lines denoting the separation from the outer myelinated portions of the axons. Interestingly, note that the collapse of the axon in Panel I presents an image similar to a yin- and yang-like icon; these were frequently found to be of this type of appearance in the transitional zones between the grey and white matter regions in the diabetic db/db models. Panel J illustrates a normal-appearing oligodendrocyte (OL) with a normal-appearing nuclear pattern of chromatin (Ch) in control non-diabetic C57BL/6J mice. Panel K depicts a marked increase in the peripheral chromatin (Chr) staining compatible with nuclear chromatin condensation that was also noted in activated microglia in the diabetic db/db models. Figures adapted with permission by CC 4.0 [33]. Magnifications are marked in the right upper portion of the each of the panels and scale bars are located in the lower left or central of these panels. M = myelin.
Oligodendrocytes, oligodendrocyte precursor cells, and myelination are not static and readily adapt to changes in their environment. The previously described alterations in myelin remodeling in the diabetic db/db models may be detrimental and could impair cognition by the slowing of transmission rates of information and action potentials in white matter tracts to distant regions. This aberrant remodeling could result in a loss of synchronicity, not only between neurons, but to entire neuronal networks [33]. Importantly, activated astrocyte foot process (end-feet) separation and retraction from neuronal synapses and myelinated neurons, as in diabetic db/db models (Figure 6 Insert 2 and Figure 7F), could interfere with the supply of energy substrates (glucose and lactate) to neurons, neuronal synapses and neuronal axons (myelinated or unmyelinated). This is especially true in regards to the astrocyte neuron lactate shuttle (ANLS) hypothesis as the astrocyte neuron coupling of the ANLS hypothesis would be interrupted due to separation and retraction [31,32,33,34].
3. PCC Effects on the Vulnerable Choroid Plexus (CP) Blood-Cerebrospinal Fluid Barrier (BCSFB)
The choroid plexus (CP) resides within each of the four ventricles and secretes and purifies the cerebrospinal fluid (CSF) that acts as a buffer to reduce traumatic brain injuries, provides nourishment, and removes neurotoxic waste. It is also important in controlling intracranial pressure, maintaining CSF ion homeostasis, and providing micronutrients vitamins, proteins and hormones for neuronal and glial development, maintenance, and function. Further, it is important in secreting numerous trophic/stabilizing proteins (brain-derived neurotrophic factor), transthyretin, and newly described choroidal phenomena, which includes the integration of circadian clock signals, immune interaction with gut microbiome, and expression of receptors to taste are now being explored even on a molecular level [35,36].
The PCCs will have a similar effect on the choroid plexus blood–cerebrospinal fluid barrier (BCSFB) as in the neurovascular unit blood–brain barrier in that they will activate the stromal fenestrated endothelial cells and the stromal cell population including macrophages, fibrocytes, and dendritic cells that reside in the space between the fenestrated capillaries and the basilar ependymal epithelial cell(s) (EPYs). EPYs have tight and adherens junctions (TJ/AJ) between adjacent ependymal cells of the choroid plexus located at the apical cell–cell interface on the cerebrospinal fluid side and are responsible for the choroid plexus barrier functions. (Figure 8) [35,36,37].
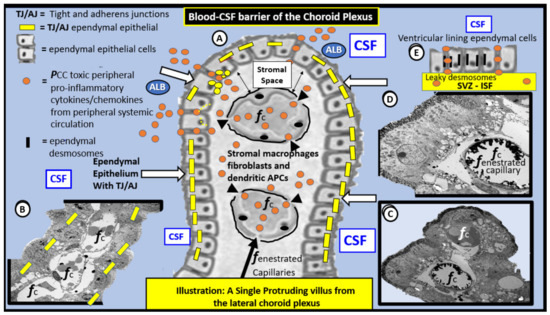
Figure 8.
Illustration and TEM images of the blood–cerebrospinal fluid barrier (BCSFB) of the choroid plexus (CP). Panel A is a hand-drawn computer-assisted illustration that depicts a single CP villus protruding into the blue background of the cerebrospinal fluid (CSF) within the lateral ventricle and note that the villus is lined by cuboidal shaped epithelial ependymal cells (white arrows) in the central portion of this image. Furthermore, note that, when these cells lose their tight and adherens junctions (TJ/AJ) (yellow bars), they allow for the escape of proinflammatory peripherally derived cytokines/chemokines (pCC) (orange spheres) or stromal cell-derived CNS CC as well as albumin (ALB) to leak into the cerebrospinal fluid CSF. Panel B demonstrates a transmission electron microscopic (TEM) morphology of a longitudinal CP villus protruding into the CSF (blue color) from a control C57BL/6J model. Note the fenestrated capillaries (fcap) within the villus and the lining ependymal epithelial cells with known TJ/AJ (inserted yellow bars). Panel C depicts a TEM cloverleaf-like image of the CP villus from the BTBR ob/ob diabetic model with marked vacuolization and vesiculation of the adjacent basilar infoldings. Note the intense electron dense staining of the microvilli at the CSF apical surface of the ependymal cells by lanthanum nitrate staining and also in Panel D which depicts the epithelial glycocalyx in intimate contact with the apical ependymal microvilli. Panel D illustrates a TEM image of the tip of the cloverleaf-like villus protruding into the CSF. Additionally, note the intense electron dense staining of fenestrated capillary (fc within the villus that is stained with lanthanum nitrate and depicts an endothelial glycocalyx) and note the important lack of lanthanum staining in the superior fcap. Panel E is an illustration that demonstrates the adjacent ependymal epithelial cells that line the entire lateral ventricles and do not have TJ/AJ; instead, they have leaky cell–cell attachments consisting of desmosomes (thin black bars) that allow molecules to move bidirectionally from the CSF to the underlying subventricular zone (SVZ) in the interstitial fluid (ISF) compartment. This leaky epithelium that lines the ventricles allow molecules to move bidirectionally from the CSF compartment to the SVZ ISF compartment based on a concentration gradient. The TEM images were acquired from the lateral ventricles of the control non-diabetic and the diabetic BTBR ob/ob model of obesity and type 2 diabetes mellitus. Illustrations in Panels A and E are not to scale, while Panels B–D have intact scale bars (5 μm in B,C and 2 μm in D) in lower left of each TEM image. It is important to note that the toxic proinflammatory cytokines and chemokines (orange spheres) are able to pass the blood–cerebrospinal fluid barrier if the TJ/AJ are rearranged, attenuated, and/or lost, in addition to their respective stromal endothelial cells and ependymal cellular receptors to the PCC dysfunctional CP. Note the key on the upper left-hand portion of this image. Inserts B, C and E adapted with permission by CC 4.0 [37].
The choroid plexus is important for many reasons including its ability to synthesize and secrete and purify the CSF, important for maintaining the intracranial pressure and maintaining ion and acidity (pH due to bicarbonate transporters). Importantly, the choroid plexus provides for micronutrient and vitamin supply, which may play a crucial role in individuals with LC/PASC since some of these patients may have a marked decreased in micronutrients, proteins, and hormones, such as leptin, thyroxin, and progesterone for neuronal and glial development, maintenance, and function [35,36,37] as a result of their acute episode with COVID-19 and lack of oral micronutrients, vitamins, and protein while in an intensive care environment or due to a lack of appetite and proper oral nutrient intake.
Importantly, the choroid plexus is more vulnerable to become injured by neurotoxins as compared to the neurovascular unit blood–brain barrier since its stromal capillaries are fenestrated and its ependymal tight junctions/adherens junctions are not as tight as compared to the brain endothelial cells of the blood–brain barrier (Figure 9) [35,36,37,38,39,40,41].
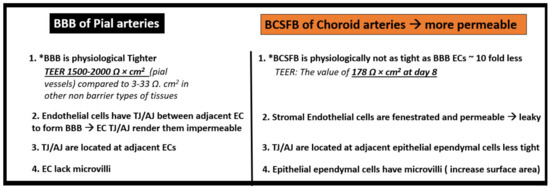
Figure 9.
Comparing the pial arterial neurovascular unit blood–brain barrier (BBB) to the choroidal arterial choroid plexus blood–cerebrospinal fluid barrier (BCSFB).
The choroid plexus contains fenestrated capillaries surrounded by a stromal space where immunocompetent macrophages, fibroblasts, and dendritic cells are located, and each of these stromal cells are capable of responding to an elevated level of PCCs and contribute to even more central nervous system-derived cytokines and chemokine (CNSCC) within the brain, since the choroid plexus blood–cerebrospinal fluid barrier, similar to the blood–brain barrier, are each neuroimmune axes [21]. Importantly, it has been recently found that the choroid plexus undergoes extensive remodeling when exposed to stress/injury such as T2DM in the diabetic BTBR ob/ob model (Figure 10) [37].
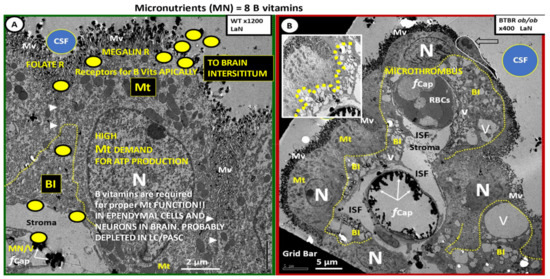
Figure 10.
Ependymal epithelial cells of the choroid plexus (CP) are remodeled in the obese diabetic BTBR ob/ob models as compared to controls. Panel A depicts the morphology of a normal non-diabetic control wild type (WT) mouse model. Note the marked presence of the highly electron dense mitochondria (Mt) in order to produce ATP to run the high-energy demands of ependymal transport systems. Importantly, note the specialized folded labyrinth structure of basilar infoldings (BI) outlined in yellow dashed lines that appear to be markedly remodeled in the BTBR ob/ob models in Panel B. In addition, note the yellow spheres that represent micronutrients/vitamins (eight B vitamins and ascorbate-vitamin C) (MN/V) transport within the ependymal cells that are eventually found within the cerebrospinal fluid (CSF) and in the CNS extracellular space. Panel B depicts the marked remodeling change in the BI (currently thought to contribute to the paracellular diffusion barrier consisting of tight and adherens junctions (TJ/AJ) of the ependymal cells of the CP), as demonstrated in Figure 6 with vacuolization and vesiculation (V and v, respectively) and marked expansion into the cytosol of the epithelial ependymal cells of the obese diabetic BTBR ob/ob models. This expansion of the BI with V and v may result in the reorganization and the attenuation and/or loss of the ependymal TJ/AJs. The V and v within ependymal cells have also been identified in traumatic brain injuries. The excessive and chronic intake of alcohol and vacuolization is also a prominent finding in ischemia and ischemia–reperfusion so the remodeling of the BIs may be a sign of early cellular damage from multiple different etiologies, as demonstrated in the obese diabetic insulin resistant BTBR ob/ob model. Additionally, note the loss of the endothelial glycocalyx and the presence of a microthrombus in the upper fenestrated capillary (fCap) where the BI demonstrate the most expansion within the ependymal cytosol. Panel B insert upper left with greater contrast and brightness to better illustrate the extensive remodeling of the BI in the BTBR ob/ob models. Both Panels A and B were stained with lanthanum nitrate to define the presence of a glycocalyx on both the capillary endothelial cells and the apical cerebrospinal fluid (CSF) surface microvilli (Mv), which significantly expand the surface area of the epithelial ependymal cells. The absent staining of the endothelial glycocalyx with extensive BI remodeling expansion may be one reason that this microthrombus has formed in the fenestrated microvessel and may also contribute to dysfunctional ependymal barrier functions with increased CNSCC within the CSF in addition to the ischemia induced locally by this microthrombus. Panels A and B were adapted with permission by CC 4.0 [33]. Adapted with permission by CC 4.0 [37]. Panel A magnification ×1200; scale bar = 2 μm and Panel B magnification ×400; scale bar = 5 μm. ISF = interstitial fluid; N = nucleus; R = receptor.
The choroid plexus has proven to be a region within the brain that is known to stain positive for ACE2 (the receptor for SARS-CoV-2) and is also known to stain positive for remanent proteins of the SARS-CoV-2 both in organoids and in autopsy case reports [42]. The choroid plexus has been long overlooked as a major barrier in the central nervous system, while the blood–brain barrier remains the primary concern for most researchers.
There are those who currently think that the choroid plexus is equally important as the blood–cerebrospinal fluid barrier since these two barriers seem to coordinate their functions, while the choroid plexus blood–cerebrospinal fluid barrier acts both as a gateway and a target [43] in allowing toxic substances, such as excessive toxic PCC and immune cells, to enter the cerebrospinal fluid and eventually into the central nervous system neuropil extracellular space containing the interstitial fluid. Interestingly, the purifying action of the cerebrospinal fluid is largely due to the finding that it turns over almost three times a day due to the highly vascular choroid plexus microvilli [35,36].
Importantly, neuroepithelial cells that line the neural tube during embryological development eventually become the ependymal lining cells of the cerebrospinal fluid ventricles. These neuroepithelial cells develop both tight and adherence junctions between adjacent ependymal cells to provide the barrier function at the paracellular location and, thus, the choroid plexus barrier function of ependymal cells are well-developed at the time of birth. Interestingly, the neuroepithelium is the original epithelial cell lining the neural tube during embryologic development. This suggests the possibility of these lining ependymal epithelial cells that originated from the neuroectoderm may become a possible regional niche or repository for a future source of stem cells later in life. It is known, for example, that the subventricular zone (SVZ) provides a rich source of progenitor cells including neurons and glial cells in adult humans. For a more in-depth examination of the embryological development of the choroid plexus, one should consider reading the paper by Liddlow [43].
During the preparation for this manuscript, an important paper was published by Yang et al [44]. This group led by Wyss-Coray demonstrated that the vulnerable choroid plexus may harbor a definite entry point of peripheral inflammation, including PCC, that are capable of signaling both neuroglial cells (microglia and astrocytes) to contribute to neuronal cell and synaptic dysfunction. Herein, their observations from their abstract are quoted below:
“We observe broad cellular perturbations which predict that choroid plexus barrier cells sense and relay peripheral infammation into the brain and show that peripheral T cells infiltrate the parenchyma. We discover COVID-19 disease-associated microglia and astrocyte subpopulations that share features with pathological cell states reported in human neurodegenerative disease 4–6. Synaptic signaling of upper-layer excitatory neurons—evolutionarily expanded in humans 7 and linked to cognitive function 8 are preferentially affected in COVID-19. Across cell types, COVID-19 perturbations overlap with those in chronic brain disorders and reside in genetic variants associated with cognition, schizophrenia, and depression. Our findings and public dataset provide a molecular framework to understand COVID-19 related neurological disease observed now and which may emerge later.”.[44]
The above references used in this quotation will also be placed in this review [45,46,47,48,49].
4. The Importance of an Endothelial Glycocalyx (ecCGx) and Epithelial Glycocalyx in Brain Barriers in Relation to LC/PASC
An intact glycocalyx of the endothelial and epithelial cells is of great importance in brain barrier homeostasis (Figure 11).
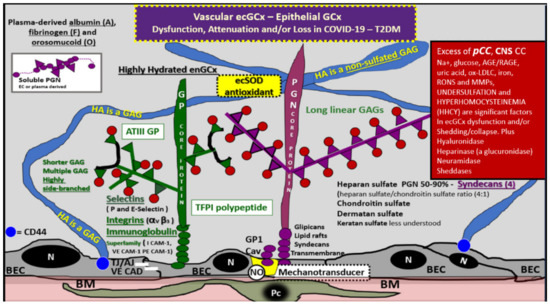
Figure 11.
Endothelial and epithelial glycocalyx in COVID-19 and type 2 diabetes mellitus. The endothelial glycocalyx (ecGCx) is the first component of the tripartite capillary blood–brain barrier (BBB) and the surface layer of choroid plexus stromal fenestrated endothelial capillaries. This illustration depicts the normal components of the ecGCx: a unique extracellular matrix. The ecGCx is composed of two classes of proteins that are mostly anchored proteoglycan(s) (PGN) (purple): glycoprotein(s) (GP) (green) and hyaluronic acid—hyaluronan (HA) (an exceedingly long polymer of non-sulfated disaccharide glycosaminoglycans (GAGs). HA (blue) may be either unattached (free floating), attached to CD44 brain endothelial cell(s) (BEC) of the plasma membrane, or form HA-HA complexes. Non-sulfated HAs not anchored to the BECs may reversibly interact at the lumen with plasma-derived albumin, fibrinogen, and soluble PGNs. The PGNs and GP side chains consist of GAGs, which are covalently bound to core proteins that are highly sulfated (red dots). The two primary PGNs are the syndecans and glypicans. The glycoproteins consist primarily of selectins (P and E), integrins (alpha v and beta 3) and the immunoglobulin superfamily of ICAM-1, VE-CAM, and PE CAM-1. Caveolae (Cav) are invaginations of lipid rafts of the brain endothelial plasma membrane (yellow invagination of BECs) and contain CD44 that importantly anchors HA to the BEC plasma membrane and glycosylphosphatidylinositol (GPI) which anchors glypican-1. The GPI/glypican-1 interaction is thought to activate endothelial nitric oxide synthase (eNOS) to produce bioavailable nitric oxide (NO) via the calcium/calmodulin-dependent Caveolin-1 (Cav-1) protein. Note the red box on right-hand side, which lists peripheral pro-inflammatory cytokines and chemokines (pCC), as well as the central nervous system cytokines and chemokines (CNSCC) in addition to numerous molecules and enzymes capable of inducing ecGCx dysfunction, collapse, or shedding. Figure adapted with permission by CC 4.0 [9,28,37]. AGE/RAGE = advanced glycation end products/receptor for AGE; ATPIII GP = antithrombin three glycoprotein; CAD = cadherin; CAM = cellular adhesion molecule; CD44 = cluster of differentiation 44; COVID-19 = coronavirus 2019 ecSOD = extracellular superoxide dismutase; GCx = glycocalyx; Ox-LDL = oxidized low-density lipoproteins; MMPs = matrix metalloproteinases; N = nucleus; Na+ = sodium; Pc = vascular mural cell pericyte(s); PECAM-1 = platelet endothelial cell adhesion molecule -1; RONS = reactive oxygen species; T2DM = type 2 diabetes mellitus; TFPI = tissue factor pathway inhibitor; VE = vascular endothelial.
The endothelial glycocalyx (ecGCx) is vasculoprotective and acts as the first barrier of the tripartite BBB and in the stromal fenestrated endothelial capillaries of the blood–cerebrospinal fluid barrier (BCSFB). Furthermore, the epithelial ependymal cells of the choroid plexus are covered by a surface layer, the epithelial glycocalyx. Additionally, the ecGCx serves as a protective coating and, thus, prevents the direct contact of the circulating blood and its constituents with the plasma membrane of the brain endothelial cells. This gel-like mesh network is responsible for multiple other protective functions in the vasculature including the promotion of vascular integrity and homeostasis, signaling blood and vessel wall capillary interactions, providing an anti-inflammatory, antithrombotic, and anti-contractile surface layer, limiting access of certain molecules based on size, steric hindrance, and especially electrostatic charge, since the glycocalyx has a strong negative net charge and is capable of repelling highly negative charged molecules and mechanotransduction of fluid shear stress and subsequent maintenance of bioavailable nitric oxide to the vessel wall. Importantly, the ecGCx is capable of modulating inflammatory processes via its prevention of leucocyte and platelet adhesion [9,24,28,37,50,51]. Furthermore, an epithelial glycocalyx is known to be present and adheres to the microvilli of the ependymal epithelial cells that line the ventricles and specifically the choroid plexus ependymal cells at the apical CSF interface (Figure 8C,D, Figure 10B and Figure 11) [37]. Further, the shedding of the glycocalyx in response to inflammatory mediators, such as cytokines and chemoattractants, was found to occur in arterioles, capillaries, and venules under various experimental models of inflammation [51].
It is also important to note that the ecGCx and epithelial glycocalyx are specialized extracellular matrices containing numerous proteoglycans and that synapses in the central nervous system are surrounded by a complex assortment of protective proteoglycans, such as chondroitin sulfate proteoglycans (CSPGs), tenascin-R (TN-R), hyaluronan (HA), and link proteins termed the perineuronal net(s) (PNNs) [52]. These PNNs are comprised of numerous compressed scaffolding of synapses and are certainly very vulnerable to the detrimental effects from proinflammatory CNSCC that are associated with increased oxidative stress (RONS) provided by the proinflammatory (activated microglia and the reactive activated astrocytes). PNNs have multiple roles but the most critical are the regulation of synaptic plasticity via stabilizing synapses and, further, provide protection of neurons and synaptic connections against damaging stressors such as the increased damaging proinflammatory CNSCCs and reactive oxygen nitrogen species that result from activation of the activated neuroglia effector cells (activated microglia and activated astrocytes) [51,52,53]. Notably, PNNs are also involved in cognition, which include encoding, maintaining, and updating memories as well as neuroglia activation by PCC and CNSCC may be involved to promote the impaired memory, focus, “brain fog”, and impaired cognition in LC/PASC [53,54].
5. Compromised Folate-Mediated One-Carbon Metabolism (FOCM) in LC/PASC: Importance of Hyperhomocysteinemia (HHCY) and Deficient Micronutrients (Vitamins B12 and B9)
Homocysteine (Hcy) is a nonessential sulfur-containing amino acid and an intermediary metabolic product derived from the demethylated essential amino acid methionine [20]. It is commonly known that plasma concentrations of homocysteine are inversely related to plasma concentrations of folate, vitamin B12, and vitamin B6, as well as to the intake of these vitamins [20,55,56,57,58,59]. Further, it is now accepted that hyperhomocyteinemia (HHCY) has vasculotoxic effects as well as neurotoxic effects that are associated with neuroinflammation, neurodegeneration, pro-oxidation, as well as proatherogenic/prothrombotic effects [20,55,56,57,58,59,60]. Elevations of Hcy or hyperhomocysteinemia (HHCY) plays an important role in the causation of oxidative stress with excess formation of reactive oxygen nitrogen species in the endothelium and in the central nervous system due to autoxidation of Hcy, formation of Hcy mixed disulfides, and interactions of Hcy thiolactones and protein homocysteinylation [20,60,61,62,63].
Accumulating knowledge has recently demonstrated that HHCY is an independent risk factor for cognitive dysfunction [59,61,62,63,64]. Both indirect and direct vascular damage can be caused by elevations in homocysteine, and HHCY has been implicated in vascular dementia with an increased risk of multiple brain infarcts and dementia as homocysteine levels rise [65,66]. Indeed, hypomethylation of DNA, proteins, and fats may render the cerebral vascular structures as well as neurons that are more vulnerable to inflammatory damage (such as PCC and CNSCC) and even apoptosis. These observations of HHCY and decreased vitamin B12, B9—folic acid, and other B vitamins emphasize the proper functioning of folate-mediated one-carbon metabolism (FOCM) typified by the methionine–folate cycle and the transsulfuration pathway. Their essential effects on cognitive diseases are of great importance as they not only apply to acute COVID-19 but, even more importantly, to LC/PASC syndrome (Figure 12) [58,59,60,61,62,64,65,66,67].
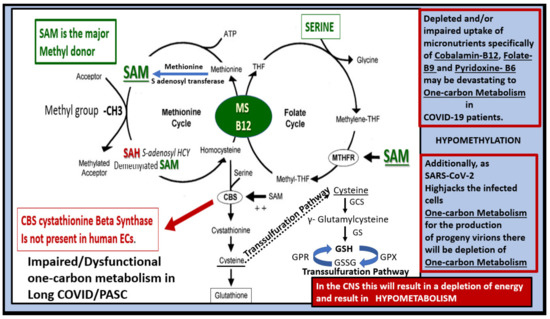
Figure 12.
Folate-mediated one-carbon metabolism (FOCM) and its important role in LC/PASC. This image demonstrates the importance of an adequate supply of methionine (an essential amino acid and an immediate precursor for S-adenosylmethionine (SAM) production), folate and the micronutrients, vitamins B12, B9 (folic acid), and how they interact with the FOCM via the donation of methyl groups. Note that SAM is the major methyl donor in this one-carbon metabolism of the methionine—folate cycle. The importance of methyl donation to the homeostasis of the CNS cannot be overestimated. Methyl donation is of great importance to neuronal function. A high demand exists for methyl group donation in the brain and limited methyl group donation will contribute to inadequate DNA methylation, histone methylation of proteins, methylation of fats (essential for proper plasma membrane function), and increased oxidative-redox stress with accumulation of reactive oxygen nitrogen species (RONS). Compromised (FOCM) will contribute to impaired neuronal dysfunction and impaired cognition. Importantly, note the transsulfuration pathway as it is so important in generating one of the cells most important antioxidants, glutathione (GSH). Indeed, methylneogenesis (de novo synthesis of methyl groups from the one-carbon pool) is necessary for proper CNS homeostasis. Therefore, the folate cycle, methionine cycle, and the transsulfuration pathway are each essential for proper FOCM in the CNS. ATP = adenosine triphosphate; CBS = cystathionine beta synthase; MTHF = methytetrahydrofolate; MS = methionine synthase; SAM = S-adenosyl methionine; SAD = S-adenosyl homocysteine; -CH3 = methyl group; THF = tetrahydrofolate; GCS = γ-glutamylcysteine synthase; GPR = glutathione-disulfide reductase; GPX = glutathione peroxidase; GS = glutathione synthetase; GSH = reduced glutathione; GSSG = oxidized glutathione.
Micronutrients consist primarily of vitamins and minerals and play an important role in CNS homeostasis. Micronutrients are an essential component of several general cellular functions in addition to proper functioning of neurologic activity, such as the synthesis of certain neurotransmitters including dopamine, serotonin, myelin formation, synaptic plasticity, and energy production, which is decreased in many neurodegenerative diseases [65,67].
When there is insufficient uptake or depletion of micronutrients, the brain cannot continue to function normally and the result will be dysfunction or disease, i.e, a loss of homeostasis, such as in the dysfunction of FOCM which occurs in both acute COVID-19 and post-COVID-19 LC/PASC [68]. Furthermore, as SARS-CoV-2 infects host cells, it high-jacks the normal one-carbon cellular metabolic machinery. This places significant demands on the host cell methyl-groups (-CH3) in order to replicate its ribonucleic acid (RNA) and provide RNA capping to produce large numbers of progeny virions. Therefore, SARS-CoV-2 infection leads to increased demands of the host cells one-carbon methyl-groups via its excess in PCC and CNSCC and the associated increase in redox-oxidative stress (reactive oxygen nitrogen species) to promote other disturbances of one-carbon metabolism. Additionally, SARS-CoV-2 may disrupt the coordination between remethylation and transsulfuration through inadequate amounts of the methyl-donor SAM. Furthermore, there may be serine depletion in addition to elevated Hcy and glutathione (GSH) depletion and the dysfunctional oxidized cofactor cobalamin (vitamin B12). Briefly, it appears that SARS-CoV-2 not only stresses the host cell one-carbon metabolism but also impairs the supply of methyl groups provided by SAM (Figure 12) [68].
Zhang et al. have been able to elegantly demonstrate that, in addition to the SARS-CoV-2 hijacking of one-carbon metabolism, it also hijacks folate (folic acid), and that folate metabolism is absolutely critical for the transfer of one-caron units for nucleotide synthesis in addition to its important role in glutathione synthesis via the transsulfuration pathway [69]. In addition, Schober et al. have demonstrated that the one-carbon pool controls mitochondrial energy metabolism via complex I and iron-sulfur clusters [70].
COVID-19 is now widely known to be associated with excess PCC and CNSCC, and this cytokine storm is associated with oxidative redox stress (reactive oxygen nitrogen species). This combination of cytokine storm and oxidative redox stress has a marked detrimental effect on the methionine cycle and its methionine synthase (MS) that converts Hcy to methionine and especially its essential cofactor vitamin B12-cobalamin. When essential cobalamin is oxidized from its functional cob(I)alamin species to the oxidized cob(II)alamin species, it will not run the MS reaction to produce methionine from Hcy and, in turn, will not be able to produce the major methyl-donor group S-adenosyl methionine (SAM) that is necessary for the host cells requirements for one-carbon metabolism.
As one begins to better understand the impaired one-carbon metabolism hypothesis in COVID-19, it becomes apparent that supplemention to patients suffering from LC/PASC with vitamin B12 and folate might have positive responses due to the impairments caused by this syndrome. For example, Regland et al. demonstrated that individuals suffering from myalgic encephalomyelitis and fibromyalgia, i.e., chronic fatigue syndrome with symptoms similar to LC/PASC, had a positive response of their symptoms when treated with injectable B12 and oral folic acid [71]. Further, accumulation of aberrant dysmorphic and dysfunctional aberrant mitochondria (aMt) in key cells including endothelial cells, pericytes, astrocyte foot processes of the neurovascular unit, protoplasmic and fibrous astrocytes, oligodendrocytes, and myelinated or unmyelinated axons, as noted in diabetic db/db models, may be related to impaired one-carbon metabolism.
6. Aberrant Mitochondria (aMt) May Contribute to Compromised Folate-Mediated One-Carbon Metabolism (FOCM) in Not Only Diabetic db/db Mice but Also May Be Present in LC/PASC
Aberrant mitochondria (aMt) were frequently identified in the diabetic db/db models in multiple cell types of the central nervous system, as previously mentioned (Figure 13).
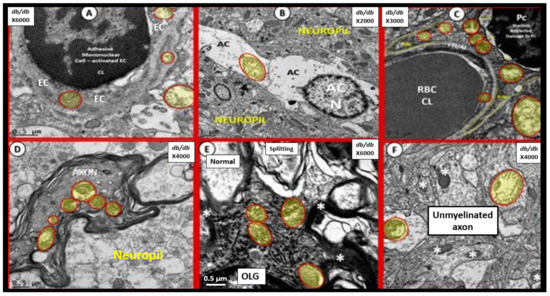
Figure 13.
Aberrant mitochondria (aMt) readily identified in multiple cell types of 20-week-old diabetic db/db Mice. Ultrastructural aMt (pseudo-colored yellow and outlined in red) were readily indentified in db/db mice in the following cells: endothelial cells (ECs) and astrocyte foot processes (ACfps) of the neurovascular unit (NVU) db/db (DBC) mice as in Panel A. Furthermore, aMt were found in protoplasmic astrocytes (ACs) in Panel B and aMt were found in pericytes (Pcs) of the NVU in Panel C. Additionally, note the aMt in the myelinated axoplasma in Panel D and the aMt found in oligodendrocytes in Panel E and the aMt of unmyelinated axons in Panel F. Magnifications ×6000; bar = 0.5 μm in Panels A and E and magnification ×2000; bar = 1 μm in Panel B and magnification ×4000; bar = 0.5 μm in Panels D,F and magnification ×3000; bar = 0.5 in Panel C. Note normal electron dense mitochondria in Panel F (asterisks). In addition, note aMt in activated microglial cells (aMGCs) in Figure 5B. Figure adapted with permission by CC 4.0 [28].
These aMt were consistently swollen with loss of electron dense mitochondrial matrix (indicating a loss of electron dense mitochondrial proteins), fragmentation of crista, loss of crista, and smudging of mitochondrial contents, as compared to normal mitochondria. Importantly, Guedj et al. have recently been able to demonstrate 18F-FDG brain PET (18F-labeled fluoro-2-deoxyglucose positron emission tomography) hypometabolism in patients with long COVID/PASC in adults [72]. They found that the regions affected included the bilateral rectal/orbital gyrus, including the olfactory gyrus; the right temporal lobe, including the amygdala and the hippocampus, extending to the right thalamus; the bilateral pons/medulla brainstem; and the bilateral cerebellum. Importantly, this hypometabolism with 18F-FDG brain PET would also be associated with not only impaired glucose uptake but also impaired cerebral blood flow and possibly regional hypoxia. Additionally, these regions may also be associated with aMt as noted in the diabetic db/db models and be associated with impaired folate-mediated one-carbon metabolism (FOCM) and hypometabolism, in addition to reflecting impaired neuronal and synaptic activity [72], since measures of regional cerebral blood flow (CBF) are also thought to reflect synaptic activity due to the tight coupling between synaptic activity, cerebral metabolic rate of glucose consumption, and CBF [73].
These aberrant mitochondria found in the brain will also importantly contribute to mitochondrial-derived oxidative and nitrosative redox stress (RONS) to cells and tissues that are affected by either decreased CBF or impaired glucose uptake due to this associated hypometabolism. Additionally, this increase in mitochondrial-derived reactive oxygen nitrogen species will contribute to the increased CNSCC, in addition to the impairment of FOCM that may invoke even further damage to neuronal and glia cells, resulting in ongoing neuroinflammation and impaired cognition related to LC/PASC.
7. COVID-19 Effects on the Hypothalamic-Pituitary-Adrenal Axis (HPA) and Adrenal Gland with Residual Effects in LC/PASC
In response to critical illnesses and marked psychogenic stress (including physical, emotional, or immunological stresses, such as excess PCC), the hypothalamic–pituitary–adrenal (HPA) axis is able to orchestrate the systemic release of cortisol in human and corticosterone in rodents referred to as glucocorticoids (generally considered anti-inflammatory immunomodulators) [74]. The brain is known to respond to peripheral systemic cues, such as inflammation and the increased PCC that occurs due to the cytokine storm in acute COVID-19 and possibly LC/PASC. While cytokines and chemokines are known to have receptors on brain endothelial cells in the neurovascular unit blood–brain barrier and the blood–cerebrospinal fluid barrier, they are also known to signal the hypothalamus early on in COVID-19 via the leaky fenestrated circumventricular organ (CVO) capillaries (Figure 14).
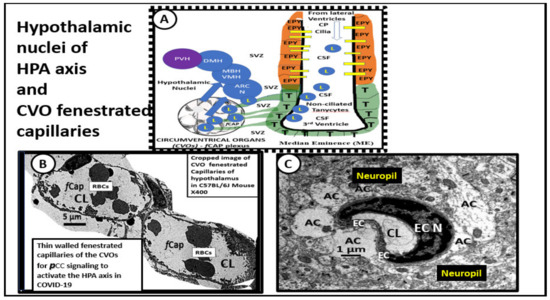
Figure 14.
Cropped images of the hypothalamic circumventricular organs (CVOs) fenestrated capillaries compared to neurovascular unit (NVU) blood–brain barrier (BBB) capillaries within the cortical hippocampal neuropil. Panel A illustrates the normal arrangement of the hypothalamic nuclei adjacent to the 3rd ventricle and how the tanycytes (T) and ependymal lining cells (EPY) of the ventricles may also be supplied by the CVO fenestrated capillaries in addition to BBB NVU capillaries in the subventricular zone of the third ventricle. Panel B depicts the phenotypic morphology of the CVOs thin-walled fenestrated brain endothelial cells (BECs) (fCap) that allow them to be leaky to the peripheral cytokines/chemokines (pCC). This arrangement allows the hypothalamus and its nuclei to be exposed to elevated systemic pCC and signal the hypothalamus to become activated. In turn, the activated hypothalamus will signal the adrenal gland to secrete increased glucocorticoids (cortisol in humans and corticosterone in rodent models), in addition to signaling the adrenal gland to produce increased epinephrin and norepinephrine. This excessive stimulation of the adrenal gland may result in adrenal fatigue (adrenal insufficiency) and/or even loss of corticosteroid production over time if the stimulation by pCC is excessive or prolonged, as in the cytokine storm in acute COVID-19 and possibly LC/PASC. Interestingly, autopsies of some COVID-19 patients have demonstrated necrosis, cortical lipid degeneration, hemorrhage and ischemic necrosis, and focal inflammation of the adrenal glands. Panel C demonstrates the thicker wall of BECs with tight and adherens junctions (TJ/AJ) in the neurovascular unit with surrounding tightly adherent and abutting astrocytes foot processes (AC) from the cortical hippocampus region in a non-diabetic control model. Both Panels A and B are from normal control mice at 20 weeks of age. These vessels have TJ/AJs and also have pericytes (not shown in this image) to complete the formation of the neurovascular unit (NVU), i.e., theblood–brain barrier (BBB) capillaries of the neuropil.
Further, stress-induced glucocorticoids are an essential response to survive severe intensive care unit (ICU) illnesses, such as COVID-19 with SARS-CoV-2, as well as the excessive emotional and burdening stresses placed on the patients in these extreme stressful situations. Indeed, this stress-induced glucocorticoid secretion in severe disease may result in a relative adrenal insufficiency state [75].
In a series of autopsy studies, Zinserling et al. have been able to demonstrate in COVID-19 decedents that the adrenal gland may be infiltrated with CD3+ and CD8+ lymphocytes in perivascular regions due to SARS-CoV-2, resulting in impaired adrenal function [76]. The lymphocytic vasculitis they described would also increase the risk of the known endotheliitis that is associated with SARS-CoV-2 and, in turn, increase the inflammatory thromboembolic state within the adrenal that has also been reported by Santana et al in their autopsy studies [77]. Santana et al. reported frequent adrenal lesions consisting of an increase in ischemic necrosis, thrombus formation, hemorrhage, and cortical lipid degeneration with these abnormalities in 12 of 28 patients autopsied [77].
Hypoadrenalism and adrenal insufficiency may be plausible in LC/PASC [75]. Siejka and Barabutis [75] have proposed at least three mechanisms for the possible development of adrenal insufficiency due to SARS-CoV-2 as follows: (i) relative adrenal insufficiency is thought to be a common condition in critically ill patients [78] and increased PCC/CNSCC in acute COVID-19 may over time result in a negative feedback toward the hypothalamic-pituitary-adrenal axis [79]; (ii) SARS-CoV-2 generates certain amino acid sequences that may mimic the hosts adrenocorticotropic hormone (ACTH) and, as a result of these corresponding antibodies, may contribute to the development of central adrenal insufficiency [75]; and (iii) the pituitary, hypothalamus, and adrenal glands each express angiotensin-converting enzyme 2 (ACE2), and SARS RNA has been identified in hypothalamic tissues of autopsied patients with COVID-19 that might cause hypophysitis and adrenalitis, and promote either secondary or primary adrenal insufficiency [75,80]. Therefore, LC/PASC patients should be carefully evaluated for adrenal insufficiency.
8. Central Nervous System Inflammatory Induced Thromboembolism: Microclots and Microbleeds in Acute COVID-19 and Possibly LC/PASC Due to Increased PCC and CNSCC
Systemic PCC and CNSCC in COVID-19 are known to induce endothelial cell activation and an inflammatory related immunothrombotic or thromboembolic state within the vasculature. This immunothrombotic state ultimately causes macro-microthrombotic complications in multiple organ systems including the central nervous system, which may relate to either macrovascular (arteries and veins ≥ 200–300 μm) or microvascular (arterioles, venules or capillaries ≤ 20 μm) thrombi or clots [81].
Microvascular thrombi may also be associated with microbleeds especially in the white matter regions. These thrombi or microbleeds associated with cerebral small vessel disease (cSVD) will result in ischemic injury and ischemia/ischemia reperfusion injury due to decreased cerebral blood flow and impaired metabolic delivery and metabolic byproduct uptake that may induce even further neuroinflammation due to increased CNSCC. This increase in CNSCC will develop in a response to injury wound healing mechanism due to activated neuroglia (activated microglia and reactive activated astrocytes) which secrete CNSCC and reactive oxygen nitrogen species, resulting in ischemia-related regional injuries due to decreased cerebral blood flow to evoke even further neurodegeneration and or impaired cognition (Figure 5 and Figure 15) [82].
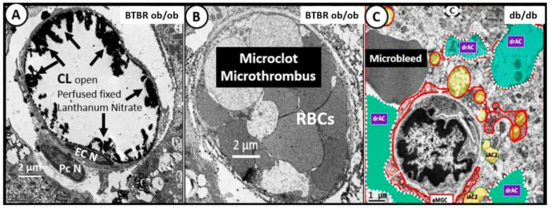
Figure 15.
Comparison of cerebral pial capillaries in cortex and choroidal capillaries in choroid plexus villus with and without microclots, microthromus, and microbleeds—microhemorrhages. This image compares a normal healthy capillary to capillaries with a microclot and a microbleed. Panel A illustrates a morphological normal open capillary within a choroid plexus villus that is perfused fixed with lanthanum nitrate to stain the endothelial glycocalyx (ecGCx) (arrows) and note that the capillary lumen (CL) is open and depicts the electron dense stained ecGCx (arrows) from a 20-week-old diabetic BTBR ob/ob model. Panel B resides within the same choroid plexus villus, as seen in Panel A; however, it does not have an ecGCx stain with lanthanum nitrate fixation and there appears to be a total occlusion of the capillary lumen with a microthrombus or microclot consisting of tightly packed adherent red blood cells, which are not usually seen when models have been perfusion-fixed prior to sacrifice, as seen in Panel A. This totally occluded capillary lumen will not allow for proper oxygenation or metabolic nutrition of surrounding tissues that will become ischemic and may result in adjacent basilar vascuolization/vesiculation in the basilar infoldings of the epithelial ependymal cell, as noted in previous Figure 10B. Panel C depicts a microbleed or hemorrhage adjacent to a dysmorphic highly remodeled capillary of the neurovascular unit of the BBB in cortical layer III of the 20-week-old db/db diabetic model with astrocyte activation detachment and retraction (drAC) (pseudo-colored cyan) and activated microglia (aMGC) (pseudo-colored red). Both the microthrombus microclot and microbleed will instigate the response to injury wound healing phase of increased inflammation and, thus, add to the pool of increased central nervous system cytokines/chemokines (CNSCC) and create a vicious cycle of micro-clot/bleeds and local inflammation. Scale bars = 2 μm in Panels A and B; 1 μm in Panel C. Figures adapted with permission by 4.0 [28,37]. EC N = endothelial cell nucleus; Pc N = pericyte nucleus.
Importantly, endothelial cell activation and dysfunction are tightly associated with cSVD and the endothelial activation in acute COVID-19 and LG/PASC with endotheliitis may certainly be playing a role with the development of microbleeds and/or clots [9,82].
Additionally, platelet activation due to endotheliitis with endothelial cell activation and dysfunction may also be a contributing factor since Pertorius et al [83] have demonstrated that, in LC/PASC, there is a persistent clotting protein pathology with increased thrombosis and an impaired fibrinolytic system resulting in impaired fibrinolysis that is associated with amyloid formation and increased antiplasmin [83,84]. Interestingly, serum amyloid A (SAA) is an amyloidogenic/fibrillogenic acute phase protein synthesized by the liver in inflammatory states (acute and chronic) that can affect coagulation by inducing amyloid formation in fibrin(ogen), as well as by inducing platelets to a more prothrombotic state [83]. Further, endothelial and platelet activation with a known endotheliitis [85] due to elevated PCC in acute COVD-19 may take a while to resolve, especially if there is an ongoing elevation of PCCs.
Recently, Chioh et al. were able to demonstrate that convalescent COVID-19 patients were susceptible to endothelial cell dysfunction due to persistent immune activation [86]. They were able to identify circulating endothelial cells in convalescent patients who had had COVID-19. Additionally, this group suggested that endothelial instability, damage, and activation/dysfunction may play a key mechanism that could be responsible for the development of post-COVID-19 vascular prothrombotic complications. Their findings, in combination with the previous discussion regarding increased inflammatory-induced thromboembolism, could also implicate an increased risk for the development to microclots and/or microbleeds, as discussed previously [81,85,86]. Further, they suggested that there may be a vicious cycle in regards to endothelial inflammation via PCC, vascular leakage, activation of the coagulation pathway, and inflammation. Certainly, the findings by Chioh et al. need to be carried out to longer time frames post-COVID-19 presentation.
9. Repurposing—Repositioning Approved Medications for LC/PASC
Our global societies have been facing the COVID-19 pandemic now for 17 months and the concerns regarding the disability of individuals with LC/PASC and their increasing numbers are of great concern. Therefore, researchers and clinicians are trying to better understand the multiple pathobiologies of LC/PASC in an attempt to be better informed as to how to treat these individuals. Indeed, repurposing or repositioning of existing medications is not a new concept [87] and one possible treatment is to suppress the chronic peripheral PCC and CNSCC neuroglia activation, neuroinflammation, and impaired cognition that may be central to the syndrome of LC/PASC. One example is to utilize anti-inflammatory modalities (such as methylprednisolone) along with the supplementation of micronutrients, such as vitamin B12 and B9–folic acid, to assist in one-carbon metabolism to treat individuals affected with this syndrome.
Importantly, Regland et al. have found that treatment with injectable vitamin B12 and oral folic acid (vitamin B9) have been successful in the treatment of patients with symptoms similar to LC/PASC who have suffered from myalgic encephalomyelitis and fibromyalgia, i.e., a chronic fatigue syndrome (ME/CFS) [71].
The author (MRH) has had his own personal experiences regarding a post-viral syndrome during the recovery from herpes simplex encephalitis and personal experiences in treating those patients with a significant post-viral syndrome due to human cytomegalic virus (hCMV) with upper respiratory infection similar to influenzae, in which affected patients were fatigued and suffered from impaired cognition who responded positively with injectable methylprednisolone and B12 and oral folic acid (B9 vitamins) (Supplementary Materials).
10. Conclusions
It has been recently estimated (May 2021) that between 10% to 35% of patients not requiring hospitalization for acute COVID-19 may develop post-COVID-19 symptoms, regardless of co-morbidities [1,16,88], while incidence rates of up to 70% have been reported among hospitalized patients and among patients with severe illnesses [89]. The true prevalence of LC/PASC syndrome is currently unknown; however, it has been stated that as many as one-in-three survivors or more depending on the depth and reliability of investigation may develop LC/PASC [89].
Some are now concerned, and rightfully so, that a significant numbers of patients who have had COVID-19 may develop myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), wherein they calculated that roughly only 10% would develop ME/CFS of approximately 25 million by the end of 2021, leaving an approximated total of 2.5 million who would meet the standards for the National Academies of Sciences, Engineering, and Medicine (NASEM) case definition criteria for ME/CFS in the United States [90]. If their projections are anywhere close to their approximated figures this occurrence, similar to the historical Spanish flu pandemic, this would indeed be another historic event in the history of chronic infection-related illness.
While the COVID-19 pandemic has caused great morbidity and mortality across the world, many have survived its acute effects and a large number of those survivors, even those with only minor acute symptoms, are now experiencing a debilitating and prolonged post-viral syndrome termed LC/PASC. This review has not only discussed and demonstrated the important role of the neuroglia cells as first responders, protectors, and gate-keepers of the CNS, but also emphasized their important role as the final effector cells in the response to injury wound healing mechanism due to the increased PCCs. Each of the neuroglial cells remodeling changes have been described in response to central nervous system (CNS) injury. Further, these neuroglial ultrastructural remodeling changes result in CNS abnormalities that are compatible with the multiple complicated neurological and neuropsychiatic symptoms associated with acute COVID-19 as well as the post-COVID-19 viral syndrome of LC/PASC. Currently, we are only at the cusp of understanding the multiple pathobiological mechanisms involved in the development of the multifactorial syndrome of LC/PASC.
The importance of increased PCCs with acute COVID that activate CNSCCs would act as an injury and a response to injury wound healing mechanism would activate neuroglia (microglial cells and astrocytes) to negatively interact with neurovascular structures, CNS neurons, and contribute to neuroinflammation and its contribution to impaired cognition was noted. The neuroglia via the activation of microglial cells and astrocytes carries out their role as a result of being the primary effector cells responsible for the CNS remodeling changes in the brain that may be associated with so many of the neurological symptoms in LC/PASC. Certainly, viremic hematogenous and neural routes for the direct viral entry into the brain still remain a strong possibility; however, at this point in time, there are only a few case reports of actually finding the virus in the central nervous system tissues or even in the cerebrospinal fluid. This may change as we continue to learn more about the direct impact of SARS-CoV-2 on the brain from various animal models and larger studies forthcoming from autopsied brain studies. One such study recently revealed that the S1 protein of SARS-CoV-2 is capable of entry into the central nervous system via the brain endothelial cells of the intact neurovascular unit blood–brain barrier by adsorptive transcytosis and that the murine angiotensin-converting enzyme2 (ACE2) is involved in brain uptake [91]. Therefore, we need to keep an open mind to future findings and incorporate them to what is currently known.
Still, it seemed overwhelming that the PCCs would be capable of causing these neurologic symptoms via activating the effector neuroglial cells and instigating an ongoing neuroinflammation. Importantly, we know that there are cytokine and chemokine receptors on the BECs of the neurovascular unit blood–brain barrier, the brain endothelial cells of the choroid plexus blood–cerebrospinal fluid barrier villous protrusions. and the ultimate epithelial ependymal barrier of the choroid plexus blood–cerebrospinal fluid barrier. This review has attempted to show that neuroglia activation by PCC and CNSCC and chronic neuroinflammation may be the common denominator for the development of the complex LC/PASC syndrome following the acute COVID-19 due to SARS-CoV-2.
Understanding the ultrastructure of the brain is important to provide a better understanding of the central nervous system’s normal and abnormal ultrastructure and how remodeling changes are associated with function, such as when studying the effects of a stressor or injury, whether it is due to central nervous systems neuronal diabetic stress/injury or understanding the stress/injury that may also be associated with the ultrastructural remodeling changes due to the stress/injury in acute COVID-19 and/or the remodeling stress/injury of LC/PASC. TEM images allow one to discern details of the brain parenchyma at micrometer and nanometer resolution and their unique features in two-dimensional images. These TEM images allow us to be able to distinguish neurons and their supportive neuroglia cells and their subcellular compartments, organelles. Cytoplasmic processes and their remodeling interactions also might occur in LC/PASC [92].
Importantly, Bernard-Valnet et al. [93] just shared (June 2021) their results from 22 patients with acute COVID-19. They examined the cerebrospinal fluid (CSF) for chemokines and concluded that their results did not indicate an active replication of SARS-CoV-2 in the CSF or massive inflammation in the CSF compartment; however, they did highlight a specific impairment of the neurovascular unit which authors felt were linked to intrathecal production of the chemokine CXCL 8. Importantly, they found that the CSF was characterized by an elevated protein and elevated albumin ratio indicative of blood–brain barrier dysfunction and leakage. Furthermore, they suggested that encephalopathies in severe SARS-CoV-2–infected patients are not due to a major viral infection of the brain. However, they did identify a strong link between encephalopathies and brain barrier impairments. This suggested that one potential mechanism could be the activation of neurovascular cells (endothelial cells, pericytes, astrocytes, and ependymal cells) which could lead to barrier disruption due to a combination of peripheral inflammation (PCC) and/or hypoxia. Further, these authors suggested a role for corticosteroid usage as a result of their findings [93].
SARS-CoV-2 has a definite brain neurotropism via its ability to bind to the angiotensin-converting enzyme 2 (ACE2) receptor on the cell membranes of neurovascular unit brain endothelial cells, neurons (both excitatory and inhibitory), and neuroglia, which include the microglia cells, astrocytes, and oligodendrocytes [94,95,96]. Additionally, hyper-activation of the immune system and an excessive aberrant cytokine storm with excessive amounts of PCC in an attempt to eradicate the SARS-CoV-2 which causes COVID-19 may trigger autoimmune responses and autoimmune disease. Multiple factors have been linked to autoimmune responses and disease, which includes genetics, age, environment, as well as viral infections including COVID-19 and now the emerging LC/PASC post-viral syndrome [97]. Autoimmunity induced by SARS-CoV-2 tropism to both peripheral organs and the central nervous system are both possible. Certain mechanisms are currently thought to be involved and include molecular mimicry between viral SARS-CoV-2 and host self-antigen epitopes, breakdown of immune tolerance (possibly due to excessive fatigue, and even burnout of the adaptive immune systems due high sustained viral loads, which result in lymphopenia and an increase in the neutrophil/lymphocyte ratio), non-specific bystander activation, epitope spreading, super-antigen presentation, and even stimulation of inflammasome platforms [94,95,96,97,98]. Once SARS-CoV-2 enters the cell via its S-1 spike protein, it will hijack the host cells organelles, nucleus, and its reproductive machinery to generate as many progeny virions as it can, while leaving the infected host cell disabled or damaged significantly and may even cause the host cell to die along with liberating a great amount of fragmented host cell self-antigens (Ag). Due to this invasion by SARS-CoV-2 and the response to injury response, there will be an influx of neutrophils initially followed by macrophages and dendritic cells of the innate immune system with even a greater amount of collateral damage to normal, non-infected local regional cells. Regardless, a high viral load will result in greater viral invasion and a greater amount of damaged fragmented molecules, and subsequent fragmented self-antigens will be liberated. These fragmented self-antigens and viral peptide antigens will set the stage for autoimmunity. Furthermore, the major histocompatibility complex (MHC)positive antigen presenting cells will present these viral antigens to the acquired immune system’s auto-reactive T-cells that will form the antibody which will then develop the antigen–antibody complex [94,99]. These possibilities should cause health care providers to become more vigilant in pursuing symptoms and complaints that may be related to the possibility of an increased emergence of autoimmune diseases as the future unfolds in the LC/PASC post-viral syndrome. In LC/PASC, the stage has been set for the development of autoimmunity, and this area of research is ripe in the knowledge to be gained. A first example of this would be the application of testing LC/PASC for autoantibodies [100] and compare those to the group who have had documented COVID-19 and recovered without developing LC/PASC.
In summary, LC/PASC is a multifactorial post-COVID-19 syndrome associated with multiple pathobiological etiologies which extend well beyond the usual time for healing and recovery. This syndrome has many parallels to myalgic encephalomyelitis/chronic fatigue syndrome with fatigue—lethargy, neurological, and neuropsychiatric symptoms. Multiple ultrastructural brain remodeling changes may be associated with this post-viral syndrome in a response to injury mechanism, as discussed in this review. Neuroglia activation and cellular remodeling, as demonstrated in the stress injury, and the response to injury wound healing mechanisms of diabetic mice models, as outlined in this review, may be similar to those who develop LC/PASC. Therefore, neuroglial activation due to increased PCC and CNSCC along with neuroinflammation might define the neurological outcomes to help explain why some post-acute COVID-19 patients develop LC/PASC syndrome due to viral infections caused by SARS-CoV-2 during this COVID-19 pandemic.
Like streams of dust emanating from a comet, LC/PASC appears to have a lasting effect from acute COVID-19 infections, and this long tail of COVID-19 is reminiscent of the long lasting 1997 comet Hale-Bopp. Unfortunately, we will continue to see the long tail of COVID-19 and will continue to observe it for some time. However, it is hoped that COVID-19 will eventually dissipate and disappear; however, we must also be prepared to co-exist with coronavirus, similar to how we have co-existed with the influenzae virus. References [101,102,103] are cited in the Supplementary Materials.
Supplementary Materials
The supplementary materials are available online at https://www.mdpi.com/article/10.3390/neuroglia2010004/s1.
Funding
Author received no funding for this project.
Institutional Review Board Statement
The tissues provided for the representative transmission electron microscopic images utilized in this manuscript were all approved in advance by the University of Missouri or the Veterans Affairs Medical Center—Puget Sound Institutional Animal Care and Use Committees, and animals were cared for in accordance with National Institutes of Health guidelines and by the Institutional Animal Care and Use Committees at the Harry S Truman Memorial Veterans’ Hospital and University of Missouri, Columbia, MO, USA or by Veterans Affairs Medical Center-Puget Sound, and care conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH).
Informed Consent Statement
Not applicable.
Data Availability Statement
Corresponding author will provide data upon reasonable request.
Acknowledgments
Author wishes to acknowledge Alexei Verkhratsky and Arthur Butt for the creation of the original Neuroglia journal. Their kind and thoughtful teaching throughout the world have made a huge difference in how we currently view neuroglia in health and neurological diseases and their important role even now through this COVID-19 pandemic. Author also wishes to acknowledge William A Banks for his support in writing this manuscript as well as supplying the novel BTBR ob/ob models that were utilized. Author also wishes to acknowledge Deana Grant of the Transmission Electron Microscopy core facility at the University of Missouri. The author especially wishes to acknowledge those who are suffering from the effects of LC/PASC—“long haulers” and how they were instrumental in getting research funded for scientists, researchers and clinicians involved in their plight and suffering.
Conflicts of Interest
The author declares no conflict of interest.
References
- Havervall, S.; Rosell, A.; Phillipson, M.; Mangsbo, S.M.; Nilsson, P.; Hober, S.; Thålin, C. Symptoms and functional impairment assessed 8 months after mild COVID-19 among health care workers. JAMA 2021, 325, 2015. [Google Scholar] [CrossRef]
- Lerner, A.M.; Robinson, D.A.; Yang, L.; Williams, C.F.; Newman, L.M.; Breen, J.J.; Eisinger, R.W.; Schneider, J.S.; Adimora, A.A.; Erbelding, E.J. Toward Understanding COVID-19 Recovery: National Institutes of Health Workshop on Postacute COVID-19. Ann. Intern. Med. 2021, 174, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S. NIH Launches New Initiative to Study “Long COVID”. National Institute of Health 23 February 2021. Available online: https://www.nih.gov/about-nih/who-we-are/nih-director/statements/nih-launches-new-initiative-study-long-covid (accessed on 29 July 2021).
- Agorastos, A.; Tsamakis, K. The need for holistic, longitudinal and comparable, real-time assessment of the emotional, behavioral and societal impact of the COVID-19 pandemic across nations. Psychiatriki 2021, 32, 15–18. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Post-COVID Conditions 8 April 2020. Available online: https://www.cdc.gov/coronavirus/2019-ncov/long-term-effects.html (accessed on 29 July 2021).
- Clark, R.A. Cutaneous tissue repair: Basic biologic considerations. I. J. Am. Acad. Dermatol. 1985, 13, 701–725. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, K.M.; Pulakat, L.; Joginpally, T.; Krueger, B.; Whaley-Connell, A.; Sowers, J.R. Possible mechanisms of local tissue renin-angiotensin system activation in the cardiorenal metabolic syndrome and type 2 diabetes mellitus. Cardiorenal Med. 2011, 1, 193–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef]
- Stroncek, J.D.; Reichert, W.M. Overview of wound healing in different tissue types. In Indwelling Neural Implants: Strategies for Contending with the In Vivo Environment; Reichert, W.M., Ed.; CRC Press: Boca Raton, FL, USA, 2008; Chapter 1. Available online: https://www.ncbi.nlm.nih.gov/books/NBK3938/ (accessed on 29 July 2021).
- Denes, A.; Thornton, P.; Rothwell, N.; Allan, S. Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain Behav. Immun. 2010, 24, 708–723. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Van Kampen, J.J.A.; van de Vijver, D.A.M.C.; Fraaij, P.L.A.; Haagmans, B.L.; Lamers, M.M.; Okba, N.; van den Akker, J.P.C.; Endeman, H.; Gommers, D.A.M.P.J.; Cornelissen, J.J.; et al. Duration and key determinants of infectious virus shedding in hospitalized patients with coronavirus disease-2019 (COVID-19). Nat. Commun. 2021, 12, 267. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Ramani, T.; Auletta, C.S.; Weinstock, D.; Mounho-Zamora, B.; Ryan, P.C.; Salcedo, T.W.; Bannish, G. Cytokines: The Good, the Bad, and the Deadly. Int. J. Toxicol. 2015, 34, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.; Pavli, A.; Tsakris, A. Post-COVID syndrome: An insight on its pathogenesis. Vaccines 2021, 9, 497. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Darley, D.; Howe, A.; Munier, C.M.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; Kelleher, A.D.; et al. Immunological dysfunction persists for 8 months following initial mild-moderate SARS-CoV-2 infection. medRxiv 2021. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and type iii interferons—Induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Low, R.N.; Low, R.J.; Akrami, A. A cytokine-based model for the pathophysiology of long COVID symptoms. OSF Prepr. 2020. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the blood–brain barriers and blood–brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Erickson, M.; Morofuji, Y.; Owen, J.B.; Banks, W.A. Rapid transport of CCL11 across the blood-brain barrier: Regional variation and importance of blood cells. J. Pharmacol. Exp. Ther. 2014, 349, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural remodeling of the neurovascular unit in the female diabetic db/db model—Part II: Microglia and mitochondria. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural remodeling of the neurovascular unit in the female diabetic db/db model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244. [Google Scholar] [CrossRef] [Green Version]
- Bouayed, J.; Bohn, T. The link between microglia and the severity of COVID-19: The “two-hit” hypothesis. J. Med. Virol. 2021, 93, 4111–4113. [Google Scholar] [CrossRef]
- Hayden, M.R. Hypothesis: Astrocyte foot processes detachment from the neurovascular unit in female diabetic mice may impair modulation of information processing—Six degrees of separation. Brain Sci. 2019, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R. Type 2 diabetes mellitus increases the risk of late-onset Alzheimer’s disease: Ultrastructural remodeling of the neurovascular unit and diabetic gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Bush, N.A.O.; Nedergaard, M.; Butt, A. The special case of human astrocytes. Neuroglia 2018, 1, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Scemes, E.; Spray, D.C. The astrocytic syncytium. In Non-Neural Cells in the Nervous System: Function and Dysfunction; Hertz, L., Ed.; Elsevier: New York, NY, USA, 2004; pp. 165–179. [Google Scholar]
- Belanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialog-Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [CrossRef]
- Shulyatnikova, T.; Verkhratsky, A. Astroglia in sepsis associated encephalopathy. Neurochem. Res. 2019, 45, 83–99. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.; Demarco, V.G. Ultrastructural remodeling of the neurovascular unit in the female diabetic db/db model—Part III: Oligodendrocyte and myelin. Neuroglia 2018, 1, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a metabolite and a regulator in the central nervous system. Int. J. Mol. Sci. 2016, 17, 1450. [Google Scholar] [CrossRef] [Green Version]
- Spector, R.; Keep, R.F.; Snodgrass, S.R.; Smith, Q.R.; Johanson, C.E. A balanced view of choroid plexus structure and function: Focus on adult humans. Exp. Neurol. 2015, 267, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Johanson, C.E.; Johanson, N.L. Choroid plexus blood-CSF barrier: Major player in brain disease modeling and neuromedicine. J. Neurol. Neuromed. 2018, 3, 39–58. [Google Scholar] [CrossRef]
- Hayden, M.; Banks, W. Deficient leptin cellular signaling plays a key role in brain ultrastructural remodeling in obesity and type 2 diabetes mellitus. Int. J. Mol. Sci. 2021, 22, 5427. [Google Scholar] [CrossRef]
- Crone, C.; Christensen, O. Electrical resistance of a capillary endothelium. J. Gen. Physiol. 1981, 77, 349–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, A.; Jones, H.C.; Abbott, N.J. Electrical resistance across the blood-brain barrier in anaesthetized rats: A developmental study. J. Physiol. 1990, 429, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Wright, E.M. Bicarbonate transport across the frog choroid plexus and its control by cyclic nucleotides. J. Physiol. 1983, 336, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Strazielle, N.; Ghersi-Egea, J.-F. Demonstration of a coupled metabolism–efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J. Neurosci. 1999, 19, 6275–6289. [Google Scholar] [CrossRef]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-csf barrier in human brain organoids. Cell Stem Cell 2020, 27, 951–961.e5. [Google Scholar] [CrossRef]
- Liddelow, S.A. Development of the choroid plexus and blood-CSF barrier. Front. Neurosci. 2015, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nat. Cell Biol. 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Author correction: Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 571, E1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigerio, C.S.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidon, A.; Zolnik, T.A.; Fidzinski, P.; Bolduan, F.; Papoutsi, A.; Poirazi, P.; Holtkamp, M.; Vida, I.; Larkum, M.E. Dendritic action potentials and computation in human layer 2/3 cortical neurons. Science 2020, 367, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.M.J.; Egbrink, M.G.A.O. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolářová, H.; Ambrůzová, B.; Šindlerová, L.; Klinke, A.; Kubala, L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediat. Inflamm. 2014, 2014, 694312. [Google Scholar] [CrossRef]
- Carulli, D.; Rhodes, K.E.; Brown, D.J.; Bonnert, T.P.; Pollack, S.J.; Oliver, K.; Strata, P.; Fawcett, J. Composition of perineuronal nets in the adult rat cerebellum and the cellular origin of their components. J. Comp. Neurol. 2005, 494, 559–577. [Google Scholar] [CrossRef]
- Reichelt, A.C.; Hare, D.; Bussey, T.J.; Saksida, L.M. Perineuronal nets: Plasticity, protection, and therapeutic potential. Trends Neurosci. 2019, 42, 458–470. [Google Scholar] [CrossRef]
- Reichelt, A.C. Is loss of perineuronal nets a critical pathological event in Alzheimer’s disease? EBioMedicine 2020, 59, 102946. [Google Scholar] [CrossRef]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.; Handy, D.E.; Castro, R. The link between hyperhomocysteinemia and hypomethylation: Implications for Cardiovascular Disease. J. Inborn Errors Metab. Screen. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, W. Significance of hyperhomocysteinemia. Clin. Lab. 2006, 52, 367–374. [Google Scholar] [PubMed]
- Graham, I.M.; O’Callaghan, P. Vitamins, homocysteine and cardiovascular risk. Cardiovasc. Drugs Ther. 2002, 16, 383–389. [Google Scholar] [CrossRef]
- Ponti, G.; Ruini, C.; Tomasi, A. Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19. Med. Hypotheses 2020, 143, 109859. [Google Scholar] [CrossRef]
- Ibrahimagić, O.; Smajlović, D.; Dostović, Z.; Vidović, M.; Tupković, E.; Kunić, S. Comment on an article: “Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19“. Med. Hypotheses 2020, 143, 110107. [Google Scholar] [CrossRef]
- Boers, G.H. Mild Hyperhomocysteinemia is an independent risk factor of arterial vascular disease. Semin. Thromb. Hemost. 2000, 26, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.J. Consequences of homocysteine export and oxidation in the vascular system. Semin. Thromb. Hemost. 2000, 26, 227–232. [Google Scholar] [CrossRef]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: Implications for atherosclerosis. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, H.P. Generation of superoxide free radical during the autoxidation of thiols. J. Biol. Chem. 1974, 249, 2151–2155. [Google Scholar] [CrossRef]
- Miller, A.L. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern. Med. Rev. J. Clin. Ther. 2003, 8, 7–19. [Google Scholar]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Pinilla, F. Brain foods: The effects of nutrients on brain function. Nat. Rev. Neurosci. 2008, 9, 568–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaddon, A.; Regland, B. COVID-19: A methyl-group assault? Med. Hypotheses 2021, 149, 110543. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, R.; Kim, S.H.; Shah, H.; Zhang, S.; Liang, J.H.; Fang, Y.; Gentili, M.; Leary, C.N.O.; Elledge, S.J.; et al. SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 2021, 12, 1676. [Google Scholar] [CrossRef]
- Schober, F.A.; Moore, D.; Atanassov, I.; Moedas, M.F.; Clemente, P.; Végvári, A.; El Fissi, N.; Filograna, R.; Bucher, A.-L.; Hinze, Y.; et al. The one-carbon pool controls mitochondrial energy metabolism via complex I and iron-sulfur clusters. Sci. Adv. 2021, 7, eabf0717. [Google Scholar] [CrossRef] [PubMed]
- Regland, B.; Forsmark, S.; Halaouate, L.; Matousek, M.; Peilot, B.; Zachrisson, O.; Gottfries, C.-G. Response to vitamin B12 and folic acid in myalgic encephalomyelitis and fibromyalgia. PLoS ONE 2015, 10, e0124648. [Google Scholar] [CrossRef]
- Guedj, E.; Campion, J.Y.; Dudouet, P.; Kaphan, E.; Bregeon, F.; Tissot-Dupont, H.; Guis, S.; Barthelemy, F.; Habert, P.; Ceccaldi, M.; et al. 18F-FDG brain PET hypometabolism in patients with long COVID. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2823–2833. [Google Scholar] [CrossRef] [PubMed]
- Jueptner, M.; Weiller, C. Review: Does measurement of regional cerebral blood flow reflect synaptic activity? Implications for PET and fMRI. NeuroImage 1995, 2, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, M.-A.; Rivest, S. The HPA—Immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front. Immunol. 2014, 5, 136. [Google Scholar] [CrossRef] [Green Version]
- Siejka, A.; Barabutis, N. Adrenal insufficiency in the COVID-19 era. Am. J. Physiol. Metab. 2021, 320, E784–E785. [Google Scholar] [CrossRef]
- Zinserling, V.A.; Semenova, N.; Markov, A.G.; Rybalchenko, O.V.; Wang, J.; Rodionov, R.N.; Bornstein, S.R. Inflammatory cell infiltration of adrenals in COVID-19. Horm. Metab. Res. 2020, 52, 639–641. [Google Scholar] [CrossRef]
- Santana, M.F.; Borba, M.G.S.; Baía-Da-Silva, D.C.; Val, F.; Alexandre, M.A.A.; Brito-Sousa, J.D.; Melo, G.C.; Queiroga, M.V.O.; Farias, M.E.L.; Camilo, C.C.; et al. Case report: Adrenal pathology findings in severe COVID-19: An autopsy study. Am. J. Trop. Med. Hyg. 2020, 103, 1604–1607. [Google Scholar] [CrossRef] [PubMed]
- Loriaux, D.L.; Fleseriu, M. Relative adrenal insufficiency. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Pastores, S.M.; Rochwerg, B.; Arlt, W.; Balk, R.A.; Beishuizen, A.; Briegel, J.; Carcillo, J.; Christ-Crain, M.; Cooper, M.S.; et al. Guidelines for the diagnosis and management of critical illness-related corticosteroid insufficiency (CIRCI) in critically ill patients (part I): Society of critical care medicine (SCCM) and european society of intensive care medicine (ESICM) 2017. Intensive Care Med. 2017, 43, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Pal, R. COVID-19, hypothalamo-pituitary-adrenal axis and clinical implications. Endocrine 2020, 68, 251–252. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Stevens, H.; Peter, K. The emerging threat of (Micro)thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Poggesi, A.; Pasi, M.; Pescini, F.; Pantoni, L.; Inzitari, D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J. Cereb. Blood Flow Metab. 2015, 36, 72–94. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in long COVID/post-acute sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. medRxiv 2021. [Google Scholar] [CrossRef]
- Page, M.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.-M.; Nell, T.A.; De Villiers, W.J.S.; De Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Chioh, F.W.; Fong, S.-W.; E Young, B.; Wu, K.-X.; Siau, A.; Krishnan, S.; Chan, Y.-H.; Carissimo, G.; Teo, L.L.; Gao, F.; et al. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. eLife 2021, 10, e64909. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, T.; Knight, M.; A’Court, C.; Buxton, M.; Husain, L. Management of post-acute COVID-19 in primary care. BMJ 2020, 370, m3026. [Google Scholar] [CrossRef]
- Becker, R.C. COVID-19 and its sequelae: A platform for optimal patient care, discovery and training. J. Thromb. Thrombolysis 2021, 51, 587–594. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Bateman, L. Will COVID-19 lead to myalgic encephalomyelitis/chronic fatigue syndrome? Front. Med. 2021, 7, 606824. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 protein of SARS-CoV-2 crosses the blood–brain barrier in mice. Nat. Neurosci. 2020, 24, 368–378. [Google Scholar] [CrossRef]
- Nahirney, P.C.; Tremblay, M.-E. Brain ultrastructure: Putting the pieces together. Front. Cell Dev. Biol. 2021, 9, 629503. [Google Scholar] [CrossRef]
- Bernard-Valnet, R.; Perriot, S.; Canales, M.; Pizzarotti, B.; Caranzano, L.; Castro-Jiménez, M.; Epiney, J.-B.; Vijiala, S.; Salvioni-Chiabotti, P.; Anichini, A.; et al. Encephalopathies associated with severe COVID-19 present neurovascular unit alterations without evidence for strong neuroinflammation. Neurol. Neuroimmunol. Neuroinflammation 2021, 8, e1029. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.-E.; Madore, C.; Bordeleau, M.; Tian, L.; Verkhratsky, A. Neuropathobiology of COVID-19: The role for glia. Front. Cell. Neurosci. 2020, 14, 592214. [Google Scholar] [CrossRef]
- Awogbindin, I.O.; Ben-Azu, B.; Olusola, B.A.; Akinluyi, E.T.; Adeniyi, P.A.; Di Paolo, T.; Tremblay, M. Microglial implications in SARS-CoV-2 infection and COVID-19: Lessons from viral RNA neurotropism and possible relevance to Parkinson’s disease. Front. Cell. Neurosci. 2021, 15, 670298. [Google Scholar] [CrossRef]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The Spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Talotta, R.; Robertson, E. Autoimmunity as the comet tail of COVID-19 pandemic. World J. Clin. Cases 2020, 8, 3621–3644. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Bartley, C.M.; Chow, R.D.; Ngo, T.T.; Jiang, R.; Zamecnik, C.R.; Dandekar, R.; Loudermilk, R.P.; Dai, Y.; Liu, F.; et al. Immunologically distinct responses occur in the CNS of COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Abou-Donia, M.B.; Lapadula, E.S.; Krengel, M.H.; Quinn, E.; LeClair, J.; Massaro, J.; Conboy, L.A.; Kokkotou, E.; Abreu, M.; Klimas, N.G.; et al. Using plasma autoantibodies of central nervous system proteins to distinguish veterans with gulf war illness from healthy and symptomatic Controls. Brain Sci. 2020, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, T.; Knight, M. Long COVID: A Primer for Family Physicians. Am. Fam. Physician. 2020, 102, 716–717. [Google Scholar] [PubMed]
- Morley, J.E. Editorial: COVID-19—The Long Road to Recovery. J. Nutr. Health Aging 2020, 24, 917–919. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).