The Identification and Interpretation of cis-Regulatory Noncoding Mutations in Cancer


Abstract
:1. Introduction
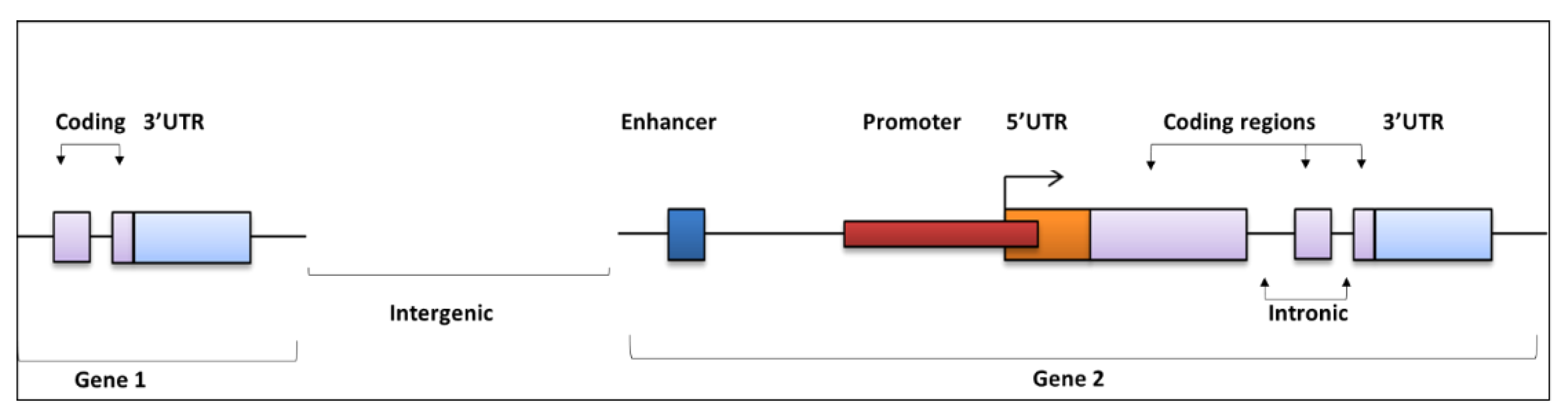
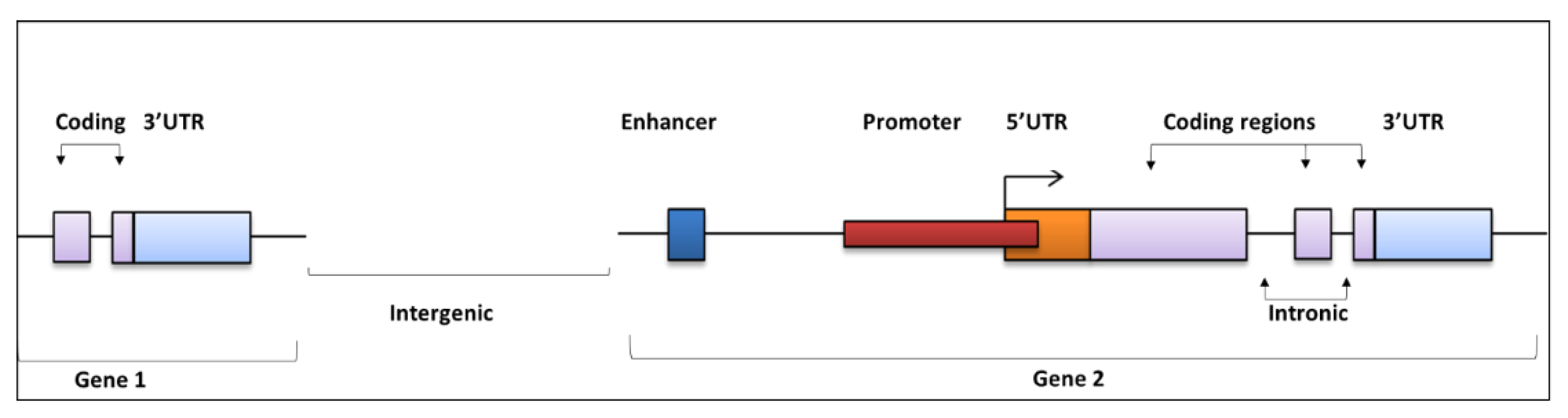
2. Regulatory Regions of the Noncoding Genome and Functional Effects
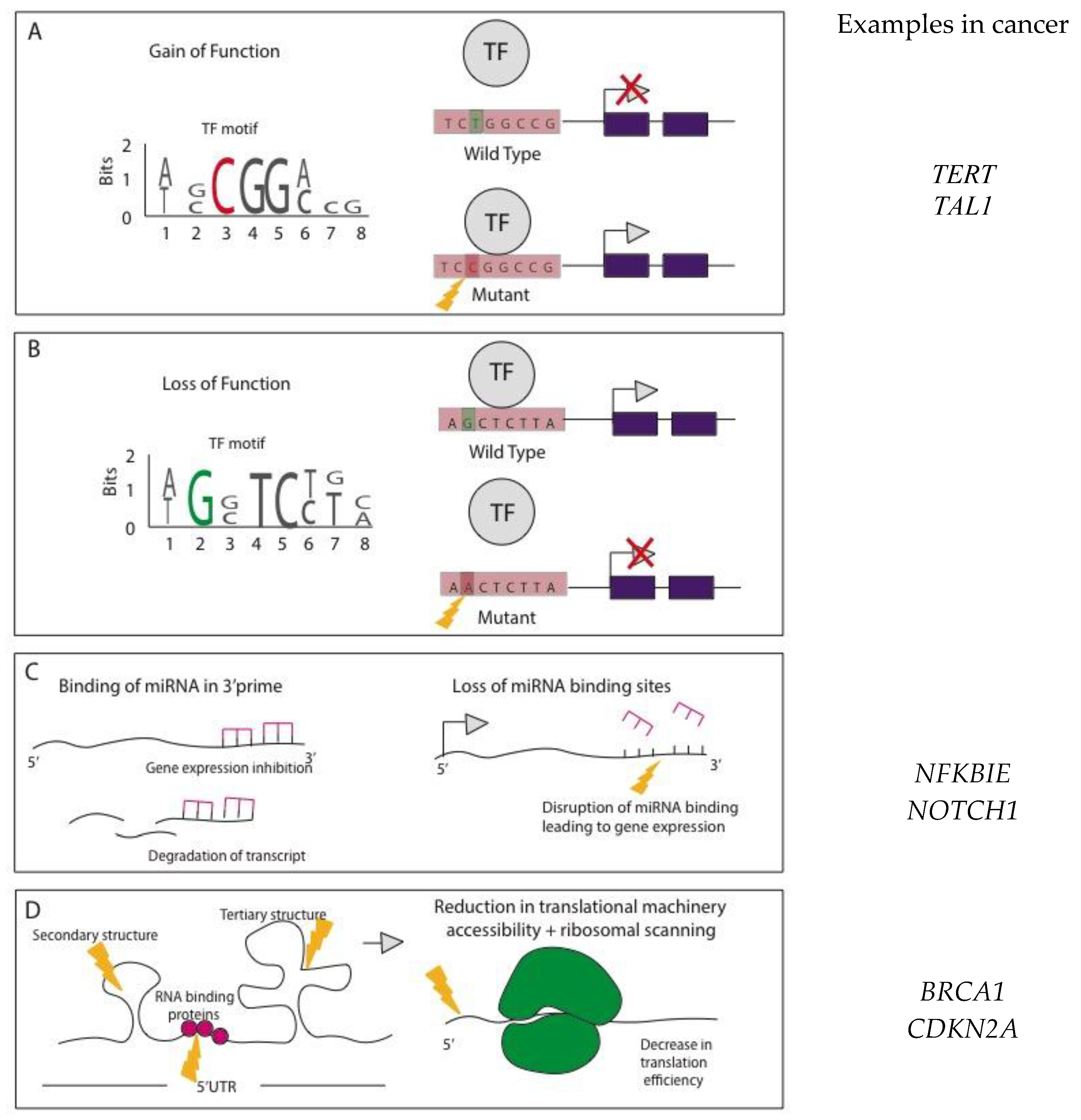
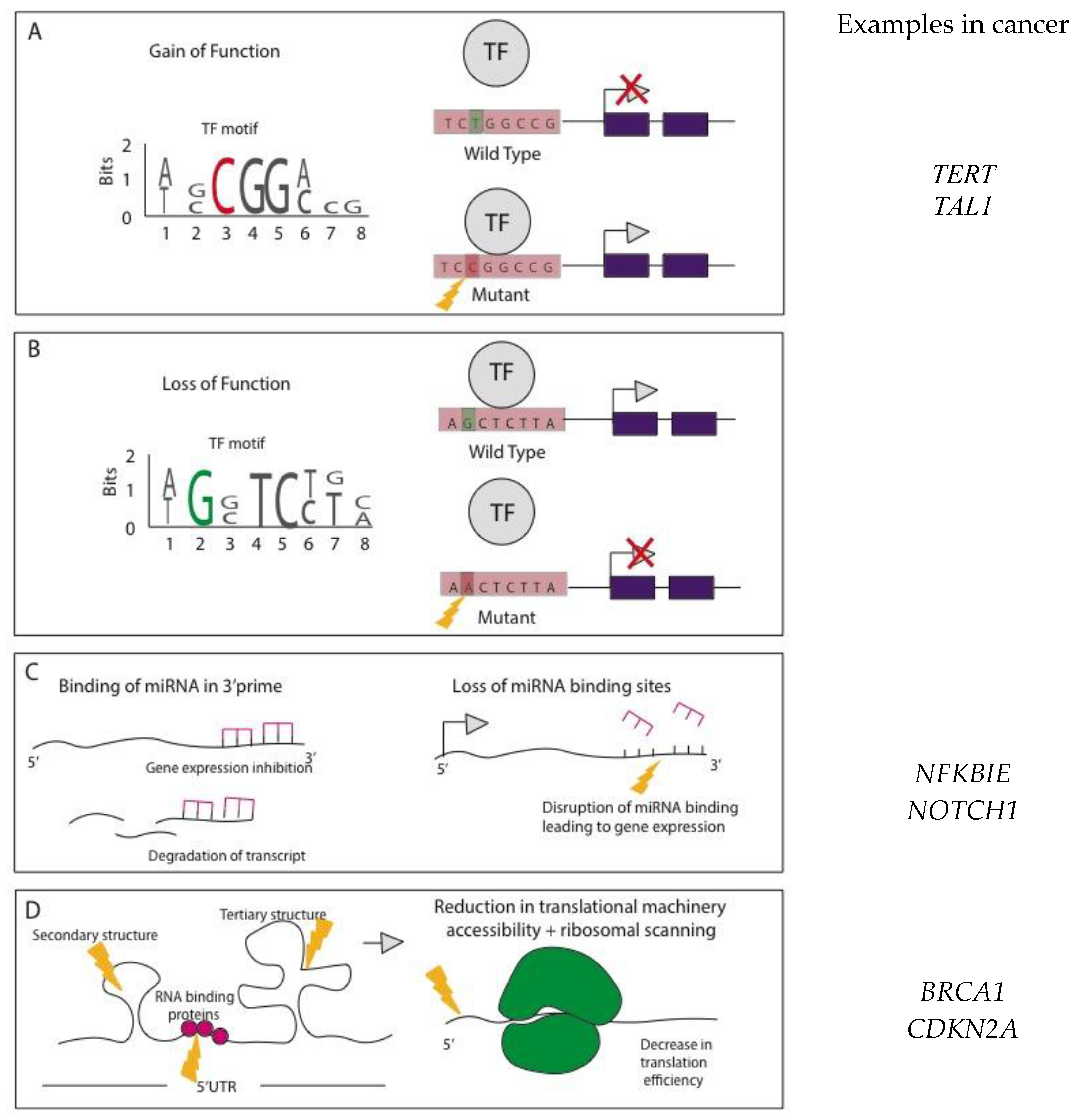
Mode of Action for NCMs
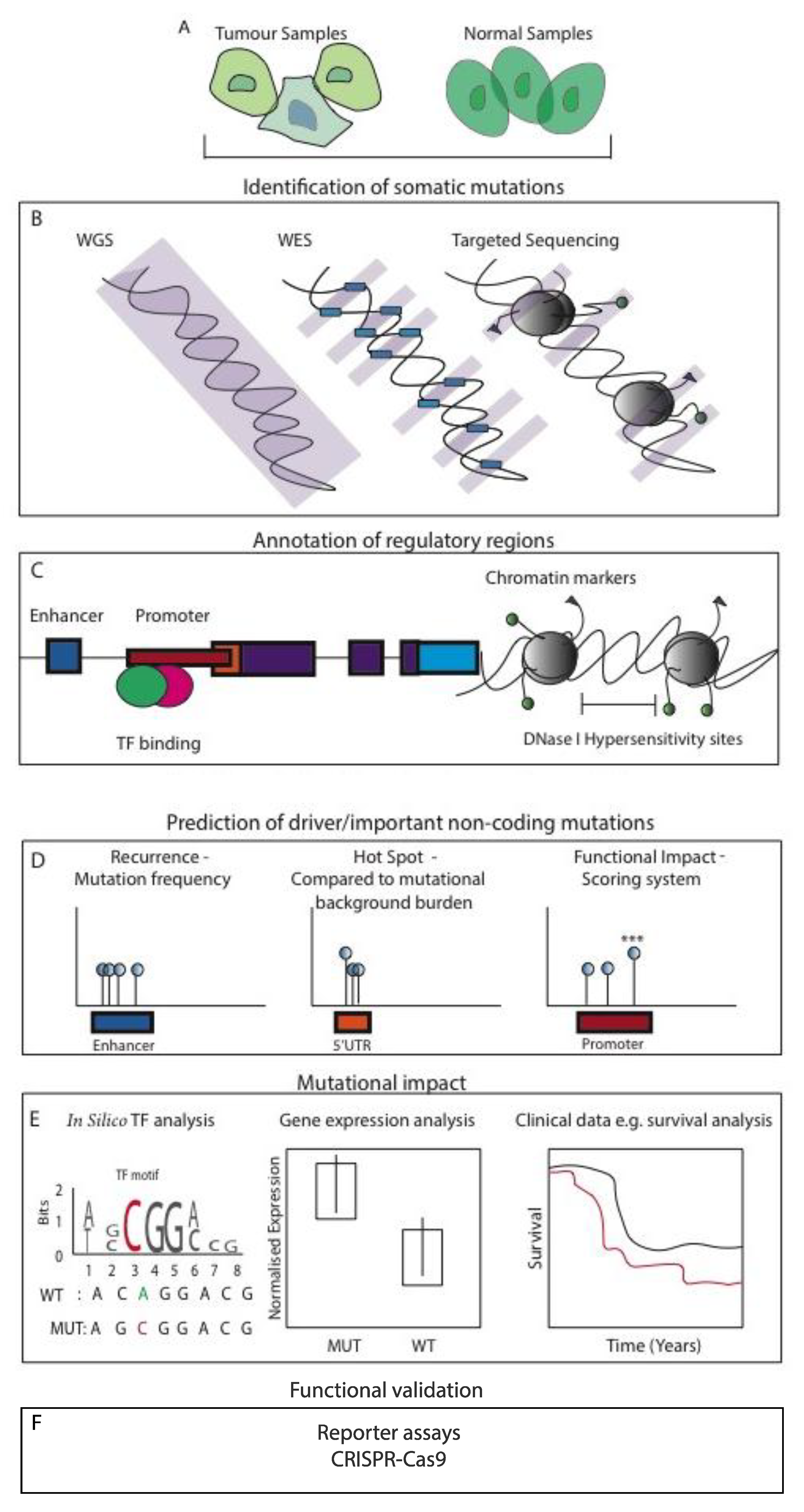
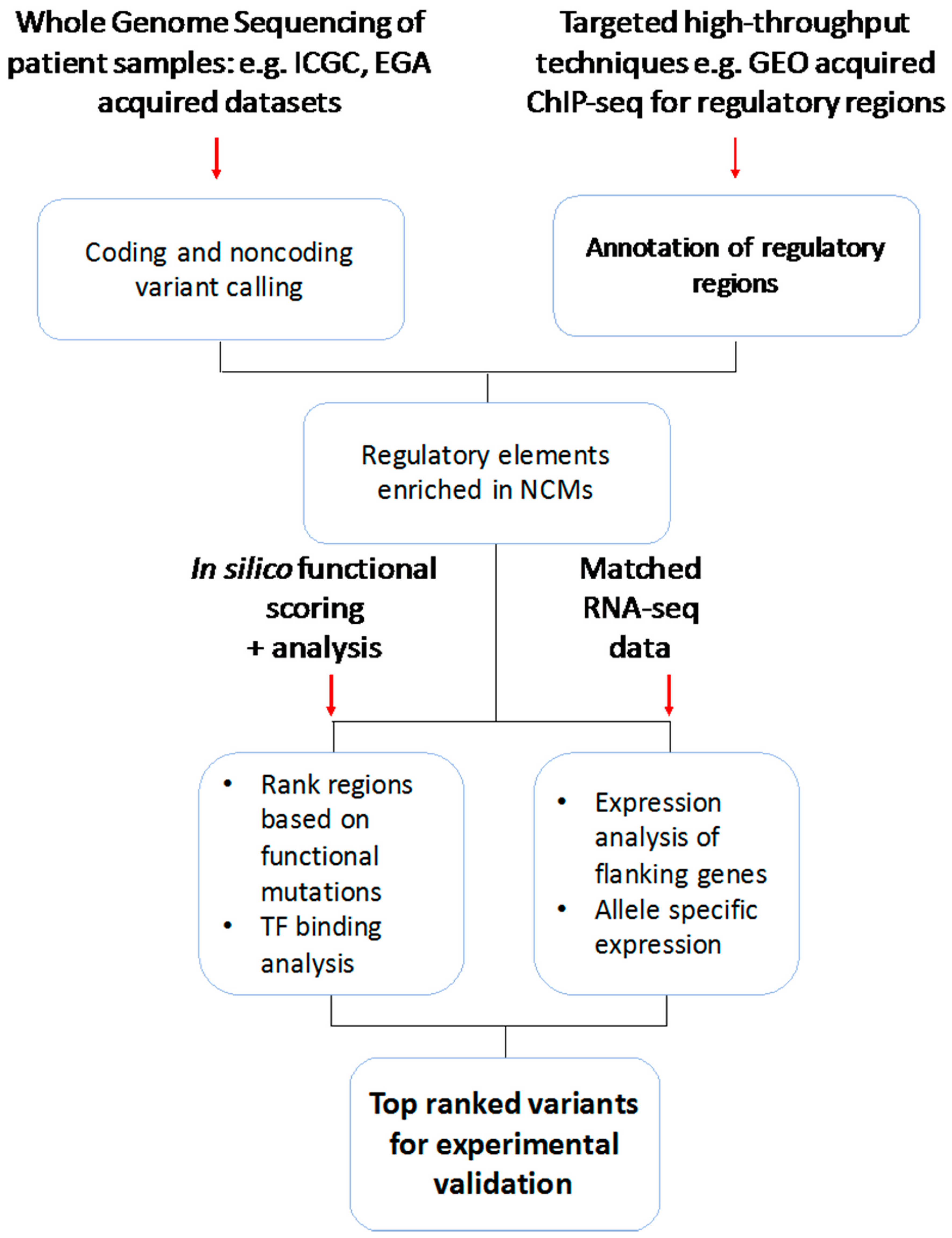
3. Noncoding Genomic Variations and Mutations Identified across Large-Scale Cancer Studies
3.1. Whole-Genome Centric Approaches
3.1.1. Whole-Genome Scans Using WGS
3.1.2. Recurrently Mutated Noncoding Clusters
3.2. Targeted and Integrative Approaches
3.2.1. Promoter-Centric
3.2.2. Active Enhancer Centric
3.2.3. Genome-Wide Chromosome Conformation
4. High-Throughput Methods and Underlying Challenges
5. Computational Resources and Techniques
6. Functional and Biological Validation of NCMs
7. Conclusions and Future Challenges
Author Contributions
Funding
Conflicts of Interest
References
- Hornshøj, H.; Nielsen, M.M.; Sinnott-Armstrong, N.A.; Switnicki, M.P.; Juul, M.; Madsen, T.; Sallari, R.; Kellis, M.; Orntoft, T.; Hobolth, A.; et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. Nature 2018, 3. [Google Scholar] [CrossRef]
- Piraino, S.W.; Furney, S.J. Beyond the exome: The role of non-coding somatic mutations in cancer. Ann. Oncol. 2016, 27, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005, 105, 2640 LP–2653 LP. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.-J.; Procter, M.L.; atland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvent chemotherapy in HER-2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Sim, E.H.; Yang, I.A.; Wood-Baker, R.; Bowman, R.V.; Fong, K.M. Gefitinib for advanced non-small cell lung cancer. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.-S.; Sakurada, A.; Cutz, J.C.; Zhu, C.Q.; Kamel-Reid, S.; Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; et al. Erlotinib in Lung Cancer—Molecular and Clinical Predictors of Outcome. N. Engl. J. Med. 2005, 353, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosoman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Romanoski, C.E.; Glass, C.K.; Stunnenberg, H.G.; Wilson, L.; Almouzni, G. Roadmap for regulation. Nature 2015, 518, 314. [Google Scholar] [CrossRef] [PubMed]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.W.; Fletcher, S.; Wilton, S.D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 2012, 69, 3613–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichellis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Bea, S.; Valdes-Mas, R.; Villamor, N.; Gutierrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Arthur, S.E.; Jiang, A.; Grande, B.M.; Alcaide, M.; Cojocaru, R.; Rushton, C.K.; Mottok, A.; Hilton, L.K.; Lat, P.K.; Zhao, E.Y.; et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat. Commun. 2018, 9, 4001. [Google Scholar] [CrossRef] [PubMed]
- Signori, E.; Bagni, C.; Papa, S.; Primerano, B.; Rinaldi, M.; Amaldi, F.; Fazio, V.M. A somatic mutation in the 5′UTR of BRCA1 gene in sporadic breast cancer causes down-modulation of translation efficiency. Oncogene 2001, 20, 4596. [Google Scholar] [CrossRef]
- Wang, J.; Lu, C.; Min, D.; Wang, Z.; Ma, X. A Mutation in the 5′ Untranslated Region of the BRCA1 Gene in Sporadic Breast Cancer Causes Downregulation of Translation Efficiency. J. Int. Med. Res. 2007, 35, 564–573. [Google Scholar] [CrossRef]
- Zhang, W.; Bojorquez-Gomez, A.; Velez, D.O.; Xu, G.; Sanchez, K.S.; Shen, J.P.; Chen, K.; Licon, K.; Melton, C.; Olson, K.M.; et al. A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat. Genet. 2018, 50, 613–620. [Google Scholar] [CrossRef]
- Li, M.J.; Yan, B.; Sham, P.C.; Wang, J. Exploring the function of genetic variants in the non-coding genomic regions: Approaches for identifying human regulatory variants affecting gene expression. Brief. Bioinform. 2014, 16, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Poursat, M.A.; Drubay, D.; Motz, A.; Saci, Z.; Morillon, A.; Michiels, S.; Gautheret, D. A Dual Model for Prioritizing Cancer Mutations in the Non-coding Genome Based on Germline and Somatic Events. PLoS Comput. Biol. 2015, 11, e1004583. [Google Scholar] [CrossRef]
- Cuykendall, T.N.; Rubin, M.A.; Khurana, E. ScienceDirect Review Systems Biology Non-coding genetic variation in cancer. Curr. Opin. Syst. Biol. 2017, 1, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Gulden, F.O.; Sestan, N. From trans to cis: Transcriptional regulatory networks in neocortical development. Trends Genet. 2015, 31, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Hosen, I.; Rachakonda, P.S.; Heidenreich, B.; Sitaram, R.T.; Ljungberg, B.; Roos, G.; Hemminki, K.; Kumar, R. TERT promoter mutations in clear cell renal cell carcinoma. Int. J. Cancer 2015, 136, 2448–2452. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef]
- Goossens, S.; Van Vlierberghe, P. Novel oncogenic noncoding mutations in T-ALL. Blood 2017, 129, 3140–3142. [Google Scholar] [CrossRef]
- Liu, L.; Dilworth, D.; Gao, L.; Monzon, J.; Summers, A.; Lassam, N.; Hogg, D. Mutation of the CDKN2A 5’UTR creates an aberrant initiation codon and predisposes to melanoma. Nat. Genet. 1999, 21, 128. [Google Scholar] [CrossRef]
- Gan, K.A.; Pro, S.C.; Sewell, J.A.; Bass, J.I.F. The Identification of Single Nucleotide Non-coding Driver Mutations in Cancer. Front. Genet. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sati, S.; Cavalli, G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma 2017, 126, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Qian, M.; Zhang, H.; Guo, Y.; Yang, J.; Zhao, X.; He, H.; Lu, J.; Pan, J.; Chang, M.; et al. Whole-genome noncoding sequence analysis in T-cell acute lymphoblastic leukemia identifies oncogene enhancer mutations. Blood 2017, 129, 3264 LP–3268 LP. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton, C.; Reuter, J.A.; Spacek, D.V.; Snyder, M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat. Genet. 2015, 47, 710. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, N.J.; Ny, L.; Nilsson, J.A.; Larsson, E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014, 46, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957 LP–959 LP. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959 LP–961 LP. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021 LP–6026 LP. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma groups Based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, P.S.; Hosen, I.; de Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426 LP–17431 LP. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Garvin, T.; Bailey, P.; Waddell, N.; Chang, D.K. Recurrent noncoding regulatory mutations in pancreatic ductal adenocarcinoma. Nat. Genet. 2017, 49, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017, 547, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.; Getz, G. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Z.; Lou, S.; Bedford, J.; Mu, X.J.; Yip, K.Y.; Khurana, E.; Gerstein, M. FunSeq2: A framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014, 15, 480. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44. [Google Scholar] [CrossRef]
- Scacheri, C.A.; Scacheri, P.C. Mutations in the noncoding genome. Curr. Opin. Pediatr. 2015, 27, 659–664. [Google Scholar] [CrossRef]
- Abraham, B.J.; Hnisz, D.; Weintraub, A.S.; Kwiatkowski, N.; Li, C.H.; Li, Z.; Weichert-Leahey, N.; Rahman, S.; Liu, Y.; Etchin, J.; et al. Small genomic insertions form enhancers that misregulate oncogenes. Nat. Commun. 2017, 8, 14385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell. 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Hume, M.A.; Barrera, L.A.; Gisselbrecht, S.S.; Bulyk, M.L. UniPROBE, update 2015: New tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2015, 43, D117–D122. [Google Scholar] [CrossRef]
- Rahman, S.; Magnussen, M.; Leon, T.E.; Farah, N.; Li, Z.; Abraham, B.J.; Alapi, K.Z.; Mitchell, R.J.; Naughton, T.; Fielding, A.K.; et al. Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood 2017, 129, 3221–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belton, J.M.; McCord, M.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the confirmation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Orlando, G.; Law, P.J.; Cornish, A.J.; Dobbins, S.E.; Chubb, D.; Broderick, P.; Litchfield, K.; Hariri, F.; Pastinen, T.; Osborne, C.S.; et al. Promoter capture Hi-C-based identification of recurrent noncoding mutations in colorectal cancer. Nat. Genet. 2018, 50, 1375–1380. [Google Scholar] [CrossRef]
- Koues, O.I.; Kowalewski, R.A.; Chang, L.W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer Sequence Variants and Transcription-Factor Deregulation Synergize to Construct Pathogenic Regulatory Circuits in B-Cell Lymphoma. Immunity 2015. [Google Scholar] [CrossRef]
- Li, G.; Cai, L.; Chang, H.; Hong, P.; Zhou, Q.; Kulakova, E.V.; Kolchanov, N.A.; Ruan, Y. Chromatin Interaction Analysis with Paired-End Tag (ChIA-PET) sequencing technology and application. BMC Genomics 2014, 15, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 10001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zook, J.M.; Salit, M. Advancing Benchmarks for Genome Sequencing. Cell. Syst. 2015, 1, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Xu, C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput. Struct. Biotechnol. J. 2018, 16, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D.; Houlahan, K.E.; Hu, Y.; Ellrott, K.; Caloian, C.; Yamaguchi, T.N.; Bare, J.C.; P’ng, C.; Waggott, D.; Sabelnykova, V.Y.; et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 2015, 12, 623. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Gratten, J. Sizing up whole-genome sequencing studies of common diseases. Nat. Gen. 2018, 50, 635–637. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [Green Version]
- Kidder, B.L.; Hu, G.; Zhao, K. ChIP-Seq: Technical considerations for obtaining high-quality data. Nat. Immunol. 2011, 12, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef] [PubMed]
- Sajan, S.A.; Hawkins, R.D. Methods for identifying higher-order chromatin structure. Ann. Rev. Genomics Hum. Genet. 2012, 13, 59–82. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epig. Chrom. 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, K.R.; Coolon, J.D.; Wittkopp, P.J. Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome. BMC Gen. 2013, 14, 536. [Google Scholar] [CrossRef] [PubMed]
- Degner, J.F.; Marioni, J.C.; Pai, A.A.; Pickrell, J.K.; Nkadori, E.; Gilad, Y.; Pritchard, J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 2009, 25, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Tavoosidana, G.; Sjolinder, M.; Gondor, A.; Mariano, P.; Wang, S.; Kanduri, C.; Lezcano, M.; Sandhu, K.S.; Singh, U.; et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 2006, 38, 1341–1347. [Google Scholar] [CrossRef]
- Harvey, C.T.; Moyerbrailean, G.A.; Davis, G.O.; Wen, X.; Luca, F.; Pique-Regi, R. QuASAR: quantitative allele-specific analysis of reads. Bioinformatics 2015, 31, 1235–1242. [Google Scholar] [CrossRef]
- Chen, J.; Rozowsky, J.; Galeev, T.R.; Harmanci, A.; Kitchen, R.; Bedford, J.; Abyzov, A.; Kong, Y.; Regan, L.; Gerstein, M. A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals. Nat. Commun. 2016, 7, 11101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dayem Ullah, A.Z.; Chelala, C. IW-Scoring: An Integrative Weighted Scoring framework for annotating and prioritizing genetic variations in the noncoding genome. Nucleic Acids Res. 2018, 46, e47. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Fricke, E.; Geffers, R.; Gossling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V.; et al. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D1284. [Google Scholar] [CrossRef]
- Mather, C.A.; Mooney, S.D.; Salipante, S.J.; Scroggins, S.; Wu, D.; Pritchard, C.C.; Shirts, B.H. CADD score has limited clinical validity for the identification of pathogenic variants in noncoding regions in a hereditary cancer panel. Genet. Med. 2016, 18, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, G.R.; Dunham, I.; Zeggini, E.; Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Schubach, M.; Jacobsen, J.O.B.; Kohler, S.; Zemojtel, T.; Spielmann, M.; Jager, M.; Hochheiser, H.; Washington, N.L.; McMurry, J.A.; et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet. 2016, 99, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Lee, D.; Gorkin, D.U.; Baker, M.; Strober, B.J.; Asoni, A.L.; McCallion, A.S.; Beer, M.A. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015, 47, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulko, B.; Hubisz, M.J.; Gronau, I.; Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 2015, 47, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.F.; Gulko, B.; Siepel, A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017, 49, 618–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionita-Laza, I.; McCallum, K.; Xu, B.; Buxbaum, J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochovsky, L.; Zhang, J.; Fu, Y.; Khurana, E.; Gerstein, M. LARVA: An integrative framework for large-scale analysis of recurrent variants in noncoding annotations. Nucleic Acids Res. 2015, 43, 8123–8134. [Google Scholar] [CrossRef]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Mercier, E.; Droit, A.; Li, L.; Robertson, G.; Zhang, X.; Gottardo, R. An integrated pipeline for the genome-wide analysis of transcription factor binding sites from ChIP-Seq. PLoS One 2011, 6, e16432. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, N.; Usvyat, D.; AC, R.M. Evaluating tools for transcription factor binding site prediction. BMC Bioinform. 2016. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rivera, A.; Santiago-Algarra, D.; Puthier, D.; Spicuglia, S. Widespread Enhancer Activity from Core Promoters. Trends Biochem. Sci. 2018, 43, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.M.; New, D.C.; Lo, R.K.; Wong, Y.H. Reporter gene assays. Methods Mol. Biol. New York, NY, USA 2009, 486, 109–123. [Google Scholar] [CrossRef]
- Dailey, L. High throughput technologies for the functional discovery of mammalian enhancers: New approaches for understanding transcriptional regulatory network dynamics. Genomics 2015, 106, 151–158. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Subtype | Study Source | Samples | Targeted Seq | WGS | WES | EXP | Chromatin Capture | ChIP-Seq | DNase-Seq | SNP-Arrays | ChIA-PET | FAIRE-Seq | Copy Number | Clinical Data | Resource | Identifier | Mutated Regions |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Melanoma | Horn et al., 2013, Science. | 169 cell lines 77 primary melanoma tumours | √ | - | - | Promoter | |||||||||||
| Melanoma | Shain, et al 2015, Nature Genetics. | 20 desmoplastic melanomas + matched normal samples | √ | √ | √ | √ | √ | Exome and targeted sequencing: Raw Microarray data | dbGaP Accession: phs000977.v1.p1. GEO: GSE55150 | Promoter | |||||||
| Breast | Rheinbay et al., 2017, Nature. | 360 primary breast cancer patients + normal | √ | √ | √ | √ | Sequencing data: | dbGAP Accession: phs001250.v1.p1. TCGA | Promoter | ||||||||
| Breast | Nik-Zainal et al., 2016, Nature. | 560 breast cancer patients | √ | √ | Raw sequencing data: | EGA: Accession EGAS00001001178 | Promoter | ||||||||||
| PDAC | Feigin et al., 2017, EMBO. | 308 PDAC patients | √ | √ | √ | WGS, Expression array, and clinical data: | ICGC AU datasets release 18 (Feb2015) | Promoter | |||||||||
| T-ALL | Mansour et al., 2014, | 2 cell lines, 8 T-ALL patients | √ | √ | √ | ChIp-seq data | GEO: GSE59657 | Super-Enhancer | |||||||||
| T-ALL | Science. Hu et al, 2017, Blood. | 31 T-ALL patients | √ | √ | √ | Sequencing data: | EGA Accession: EGAS00001001858 EGAS00001002172 | Intronic, Enhancer, Promoter | |||||||||
| CLL | Puente, et al., 2015, Nature. | 452 CLL patients + 54 MBL | √ | √ | √ | √ | √ | √ | √ | √ | Sequencing, expression and genotyping array data: | EGA Accession: EGAS00000000092 | UTR, Enhancer | ||||
| Colorectal | Orlando et al., 2018, Nature Genetics. | 19,023 promoter fragments from cell lines | √ | √ | √ | √ | √ | Hi-C, CHi-C, ChIP-seq sequencing: TF ChIP-seq: Survival data: | EGA: EGAS00001001946 GEO: GSE49402 GEO: GSE33113, GSE39582 | Enhancer | |||||||
| B-cell Lymphoma | Koues et al., 2015, Cell | Purified malignant B-cells from 18 FL patients | √ | √ | √ | All data: RNA-seq, Array, ChIP and FAIRE-seq: | NCBI Gene Expression Omnibus: GSE62246 | Enhancer | |||||||||
| DLBCL | Arthur et al, 2018, Nature Comm | 153 DLBCL tumour/norm pairs | √ | √ | √ | √ | √ | √ | 146 WGS sequence data: 1001 WES sequence validation data: | EGA: Accession EGAS00001002936 EGAS00001002606 | 3’UTR | ||||||
| Liver | Fujimoto et al., 2016, Nature Genetics | 300 Liver Cancer Patients | √ | √ | √ | Sequencing data: Mutation data: | EGA. Accession: EGAD00001001881, EGAD00001001880, EGAS00001000671, ICGC database release 18 (Feb 2015) | Promoter/Enhancer |
| High-throughput Technology | Function | Pros | Caveats | Ref |
|---|---|---|---|---|
| WGS | Identify mutations genome wide |
|
| [63,70] |
| WES | Identify mutations within exon regions. |
|
| [70] |
| ChIP-seq | Targeted approach to identify NCMs in putative functional regulatory regions. |
|
| [71] |
| DNase-seq | The identification of DNase I hypersensitivity site, mapping open chromatic genome wide. |
|
| [72,73] |
| ATAC-seq | Mapping chromatin accessibility genome-wide using a Tn5 transposase which inserts adaptors into regions of open chromatin |
|
| [74,75] |
| FAIRE-seq | Allows the identification of nucleosome depleted regions, mapping regions of open chromatin. |
|
| [75,76] |
| RNA-seq | Measure of gene expression. |
|
| [77,78] |
| 4C-seq | Identification of long-range DNA contacts with a single genomic locus of interest. |
|
| [79] |
| Hi-C-seq | Identification of long-range chromatin interactions on a global level. |
|
| [31] |
| ChIA-PET | A combination of ChIP and 3C techniques allowing the analysis of both protein-DNA complexes and long-range interactions, genome wide. |
|
| [31,61] |
| Computational Analysis Methods | Resources/Software | Method | References |
|---|---|---|---|
| Regulatory annotation resources | ENCODE | ChIP-seq, DNase-seq, ATAC-seq, Hi-C | [10] |
| Roadmap Epigenomics | ChIP-seq, DNA Methylation, RNA-seq | [11] | |
| FANTOM Consortium | CAGE | [12] | |
| Functional Scoring | CADD | Machine-learning algorithm | [88] |
| GWAVA | [89] | ||
| FATHMM-MKL | [90] | ||
| Genomiser | [91] | ||
| DeepSEA | Directly learn sequence codes from ENCODE annotations | [92] | |
| DelaSVM | [93] | ||
| FitCons | Selective pressure and divergence | [94] | |
| LINSIGHT | [95] | ||
| FunSeq2 | Weighted scoring system | [48] | |
| Eigen | [96] | ||
| IW-scoring | [83] | ||
| Regulome DB | Heuristic Scoring | [35] | |
| Rate based methods with incorporated background mutation analysis | MutSigNC | [46] | |
| LARVA | [97] |
| Traditional Reporter Based Assay | Source of DNA | Size of Test DNA Fragment | Analysis | Detection Method |
|---|---|---|---|---|
| Luciferase/GFP based reporter assays | DNA template from arbitrary source to amplify with designed primers | ~1.5–2 kb | Enhancer + promoter | Luciferase activity (luminator) or GFP activity (quantitative cytometry) |
| High-throughput reporter assays | ||||
| MPRA CRE-seq | Microarray synthesis of DNA sequences | 200–300 bp | Enhancer + promoter | RNA-sequencing |
| STARR-seq | Sheared DNA from arbitrary sources | 1–1.5 kb | Enhancer discovery (also including intergenic and intronic regions) | RNA-sequencing |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, M.B.; Wang, J. The Identification and Interpretation of cis-Regulatory Noncoding Mutations in Cancer. High-Throughput 2019, 8, 1. https://doi.org/10.3390/ht8010001
Patel MB, Wang J. The Identification and Interpretation of cis-Regulatory Noncoding Mutations in Cancer. High-Throughput. 2019; 8(1):1. https://doi.org/10.3390/ht8010001
Chicago/Turabian StylePatel, Minal B., and Jun Wang. 2019. "The Identification and Interpretation of cis-Regulatory Noncoding Mutations in Cancer" High-Throughput 8, no. 1: 1. https://doi.org/10.3390/ht8010001