1. Introduction
Cystinuria is a rare genetic disorder that causes a defect in the transporter of dibasic amino acids across membranes in the proximal renal tubule[
1,
2]. Affected patients have a defect in the reabsorption of filtered dibasic amino acids and therefore excrete large amounts of cystine, arginine, lysine, and ornithine in their urine. Cystine is poorly soluble at physiological urine pH and crystallizes in the urinary tract, causing recurrent cystine renal calculi[
1,
3]. As the other amino acids (arginine, lysine, and ornithine) are more soluble in urine, there are no adverse clinical consequences of high urinary excretion.
The dibasic amino acid transporter is a heterodimer composed of 2 subunits (encoded by SLC3A1 and SLC7A9) joined by a disulphide bridge[
4,
5]. There is a high prevalence of mutations in either subunit gene, or more rarely, both genes. Biallelic mutations of SLC3A1 are classified as Type A cystinuria. SLC3A1, located on chromosome 2, encodes the heavy subunit of the dibasic amino acid transporter, and inheritance is autosomal recessive with 100% penetrance[
6,
7]. Heterozygotes do not have an elevated risk for nephrolithiasis, and urinary excretion of cystine is usually within the normal range. Some patients with duplications of exons 5–9 of SLC3A1 may however excrete increased levels of urinary cystine and form cystine stones[
8]. Biallelic mutations of SLC7A9 are classified as Type B cystinuria. SLC7A9 is located on chromosome 19 and encodes the light subunit of the amino acid transporter[
6]. Heterozygotes may excrete increased levels of cystine and rarely form stones. Type B cystinuria therefore has either an autosomal recessive inheritance pattern or is autosomal dominant with incomplete penetrance[
7,
8]. Bigenetic mutations of both SLC3A1 and SLC7A9 rarely occur and are referred to as Type AB cystinuria.
Although cystinuria is a rare condition, patients develop stones at a young age and have an extremely high stone recurrence rate and a high risk for the development of chronic kidney disease[
8,
9,
10].
There are population-dependent variations in the proportion of type A and B genotypes, with an equal distribution in the American population and a higher preponderance of type A genotype in the United Kingdom, France, and Eastern Europe. There is a preponderance of type B genotype in cystinuria patients in Spain[
11]. According to the Human Gene Mutation Database, there are 257 SLC3A1 and 170 SLC7A9 mutations identified[
12]. It is uncertain whether cystinuria patients in South Africa carry known mutations noted in other countries or whether novel mutations exist in our population.
Although outcomes have been reported to be worse in male patients, clinical phenotype-genotype correlations have not been reported. Outcomes and management for both Type A or B cystinuria are similar[
1].
There are no publications reporting on cystinuria patients in South Africa, therefore nothing is known about the local prevalence, pathology, or genetics of cystinuria. Knowledge of the local mutations in South Africa could contribute to the development of a Sanger sequencing technique for the most common local mutations to provide a cost-effective locally accessible test. Considering South Africa’s colonial history, a “founder effect” mutation was considered possible, and therefore a predominant SLC3A1 or SLC7A9 mutation was expected.
This study aims to describe the genetic mutations and clinical phenotype (age at presentation, number of stone episodes, number of stone procedures) in the first South African cohort of patients with cystinuria.
2. Methods
Our unit is a general metabolic stone clinic at a tertiary referral center, Groote Schuur Hospital in Cape Town, South Africa. Patients are followed up every 3 months, every 6 months, or annually depending on their renal stone type and clinical status. Quantitative urinary amino acid measurement is done for all patients diagnosed with cystinuria on stone analysis or if clinical factors raise suspicion for cystinuria. Patients with cystinuria based on either stone analysis or elevated urine cystine excretion were identified from an existing stone registry (R003/2018) and were invited to participate. All patients with confirmed cystinuria who were willing to consent were included.
A single saliva sample or 2-cheek swabs were collected between September 2021 and December 2021 using a standard test kit provided by Invitae laboratories for DNA sequencing. Samples were packaged and shipped to Invitae laboratories. Genomic DNA was enriched using a hybridization protocol and sequenced using Illumina technology focusing on the coding exons and flanking intronic sequences. Copy number variations (deletions and duplications of exons) were assessed using Invitae laboratories in-house algorithm to determine copy number at each target by comparing read depth for each target in the proband sequence with mean read-depth and read-depth distribution from a set of clinical samples. In cases where a copy number variation was identified, multiplex ligation-dependent probe amplification (MLPA) was done to confirm the variant.
Chart reviews and patient interviews were done to record patient demographics (age, gender), patient medical history (other illnesses, treatments for cystinuria), family history (relatives with calculi, relatives with “kidney problems,” relatives with known cystinuria, consanguinity), stone-specific history (type, location, age at first presentation, current stones), complications of cystinuria (renal failure, nephrectomy, chronic kidney disease, dialysis), previous investigations (24-hour urine collection, urine cystine:creatinine ratio, stone analysis), number of lifetime stone events, and number of stone procedures. Patient weight and height were also measured.
Continuous data was expressed with the appropriate measures of central tendency. Categorical data was reported as percentages and proportions. IBM SPSS version 27 was used for all analyses. This protocol was reviewed by UCT Human Research Ethics Committee (HREC REF: 215/2021). All patients completed informed consent before participation in the study.
3. Results
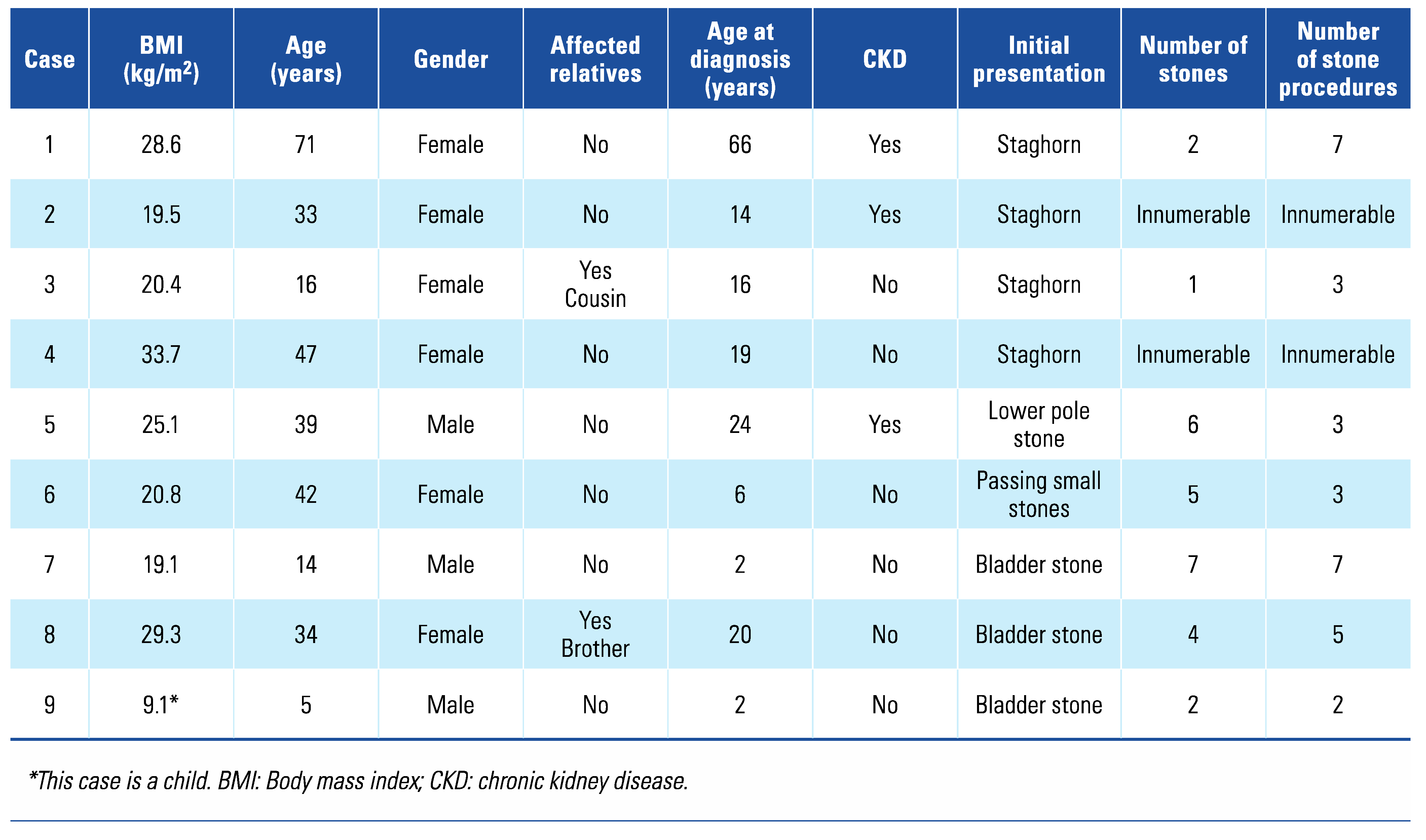
Of the 12 patients identified for possible inclusion in the study, 9 patients were included; 1 patient died of renal failure at the age of 83 years a month prior to the initiation of this study, 1 patient could not be traced, and 1 patient declined to participate. The mean age (± SD) of patients was 33.43 ± 19.96 years. Three male patients and 6 female patients were included. The median age (± IQR) at initial diagnosis of cystinuria was 16 ± 18 years, but the age ranged from 2 to 66 years. Three of 9 patients included (33.3%) had chronic kidney disease (CKD); however, none were receiving dialysis. The number of previous stone procedures per patient was difficult to assess, as 2 patients reported “too many to remember.” As both received treatment at multiple sites, the exact number of previous procedures could not be identified for either of these patients. For the other 7 patients, the number of stone episodes varied from 1 to 7 and the number of procedures from 3 to 7. Most patients initially presented with a staghorn calculus (4/9; 44.4%). None of the patients reported consanguinity. One patient reported a cousin with renal calculi since infancy, but the diagnosis of cystinuria could not be confirmed in this relative. One patient reported a brother known with cystinuria and recurrent renal calculi since teenage years; however, he declined to participate in the study (
Table 1).
The mean serum creatinine (± SD) was 84 ± 38 µmol/L. The mean urine cystine (± SD) was 2083 ± 1249 nmoL/mg creatinine; however, results were only available for 6 patients. One result was reported as “very high.” It is unclear why it was not reported as a numerical value. As the result was in keeping with the clinical picture and a stone analysis confirmed cystine nephrolithiasis, urine cystine measurement was not repeated for this patient.
Eight patients had mutations in the SLC3A1 gene; 1 patient had mutations in both SLC3A1 and SLC7A9. Of the patients with only SLC3A1 mutations, 1 patient was homozygous and the rest were compound heterozygotes (two different mutations identified in the same gene). Four patients had two pathogenic mutations in SLC3A1, and 3 patients had a pathogenic variant in addition to an “uncertain significance” variant in SLC3A1 (
Table 2).
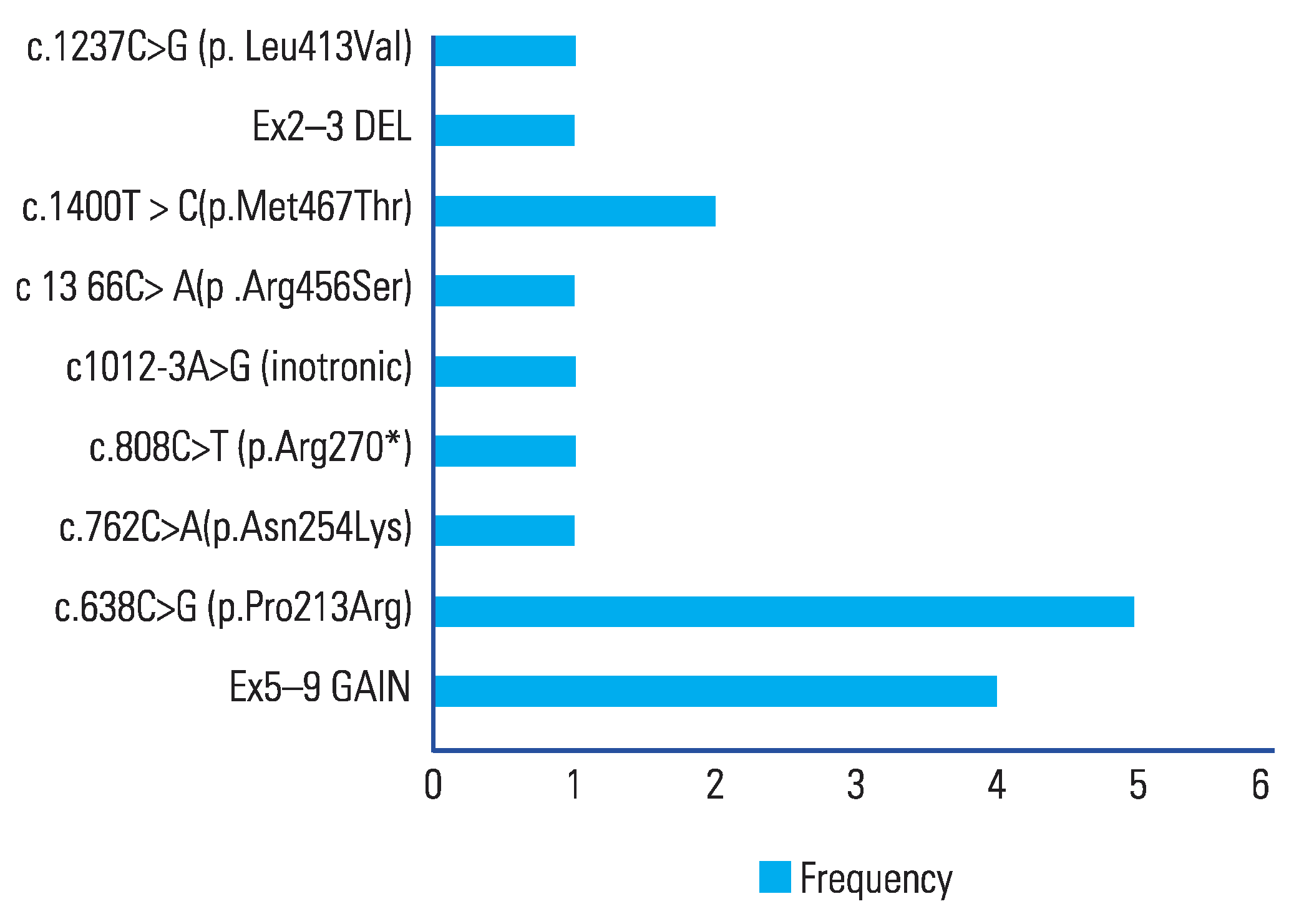
There were 5 pathogenic variants identified in SLC3A and 4 variants of “uncertain significance” reported. The most common mutations in SLC3A1 was c.638C>G (p.Pro213Arg) in 5/9 (55.5%) patients. The second most common was duplication of exons 5–9 (copy number = 3) seen in 4/9 (44.4%) patients (
Figure 1) The SLC3A1 variant c.638C>G (p.Pro213Arg) was reclassified from “uncertain significance” to “pathogenic” by Invitae laboratories during the course of this study.
Although most of the mutations identified were either present in population databases or had been previously observed in other individuals with cystinuria who submitted samples to Invitae laboratories for genotyping, there were two novel mutations identified. The novel mutation c.762C>A (p.Asn254Lys) in SLC3A1 and c.1402C>T(p.Pro468Ser) in SLC7A9 were both identified in the same patient who also had another known pathogenic mutation in SLC3A1 (
Table 2).
4. Discussion
This is the first single-population study of genetic mutations in cystinuria patients in Africa. There was marked heterogeneity in both genotype and phenotype, as has been described in other populations[
5,
6,
8,
10,
13].
We have reported two novel mutations, one each in SLC3A1 and SLC7A9 in a single patient (case 7). Although both novel mutations are of unknown significance, the patient also has a heterozygous pathogenic mutation of SLC3A1 and a clear clinical history of recurrent cystine calculi since infancy. It is therefore likely that one or both mutations are pathogenic and that the patient is a double compound heterozygote.
Three patients (cases 2,3,9) with SLC3A1 mutations were compound heterozygotes with one pathogenic mutation and one “unknown significance” mutation. Considering the clear cystinuria phenotype (based on recurrent cystine renal stones and elevated urine cystine excretion) in all 3 cases, the mutations classed as “uncertain significance” are extremely likely to be pathogenic.
A pathogenic mutation of SLC3A1 [c.638C>G (p.Pro213Arg)], not present in population databases, was the most common mutation reported in this series. There were 9 mutations (5 pathogenic and 4 “unknown significance”) in SLC3A1 and 1 in SLC7A9. Two of these were novel mutations.
The predominance of SLC3A1 mutations in this population is similar to that of populations from the United Kingdom, France, and Eastern Europe[
11,
13]. The exon 5–9 duplication in SLC3A1, which was one of the most common mutations in this cohort (4/9; 44.4%), was the most common mutation encountered in a large UK series (24/88; 27%)[
13]. Considering that South Africa was a former English and Dutch colony, we postulated that a single founder gene could be identified in this series. No predominant founder gene could be identified. Although there was an overwhelming predominance of SLC3A1 mutations, there were many different types of SLC3A1 mutations and most of the participants were compound heterozygotes.
This study was limited by the small number of patients included and the incomplete urine cystine results for the group. These limitations restricted the potential for genotype-phenotype correlations in the series. No other patients could be identified for recruitment at three other state-funded healthcare services, both within the region and beyond. Limited access to renal stone analysis and metabolic renal stone services in the region could lead to underdiagnosis of cystinuria in South Africa and on the rest of the continent. The lack of inclusion of family members for genotyping and phenotyping in cases of variants of unknown significance limited the assessment of variant significance to clinicopathological correlation alone.
In the future, recruitment of patients from privately funded healthcare services and founding a national or African registry for rare stone diseases could enable broader recruitment for a large-scale South African or African study. This would facilitate collaboration and data sharing with international groups to allow for pooling of cohorts to glean further insights into this rare condition.
5. Conclusion
This “first in Africa” series of cystinuria patients showed marked heterogeneity in both phenotype and genotype with a predominance of SLC3A1 mutations. The heterogeneity in genetics and clinical features seems similar to findings reported in populations in Europe with cystinuria.


{kind=link}