
Integrating Embeddings from Multiple Protein Language Models to Improve Protein O-GlcNAc Site Prediction



Abstract
:1. Introduction
2. Results
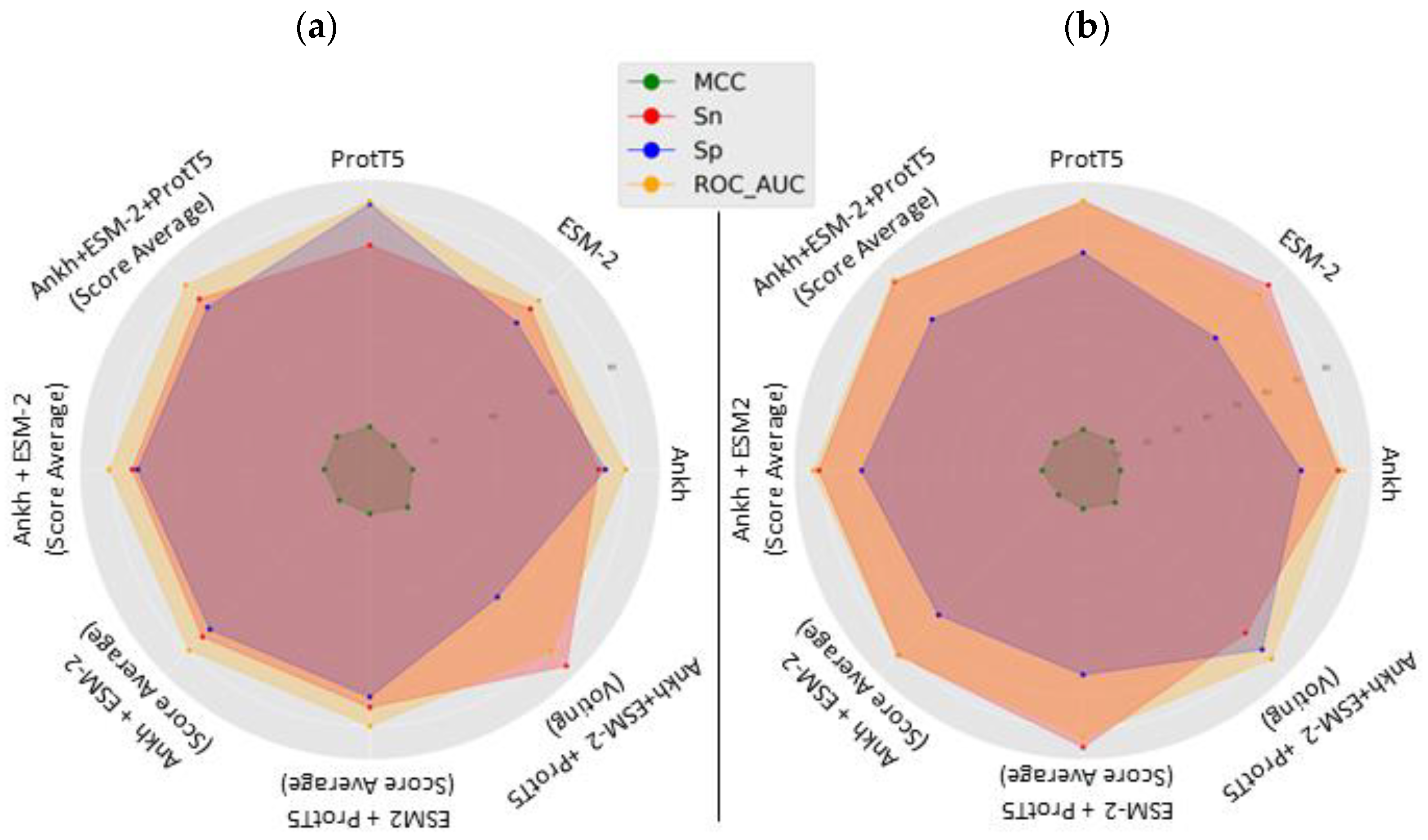
2.1. Comparison with Existing Tools
2.2. Case Study: Predicted Sites for O-GlcNAcylation in Human Galectins
3. Discussion
4. Materials and Methods
4.1. Dataset
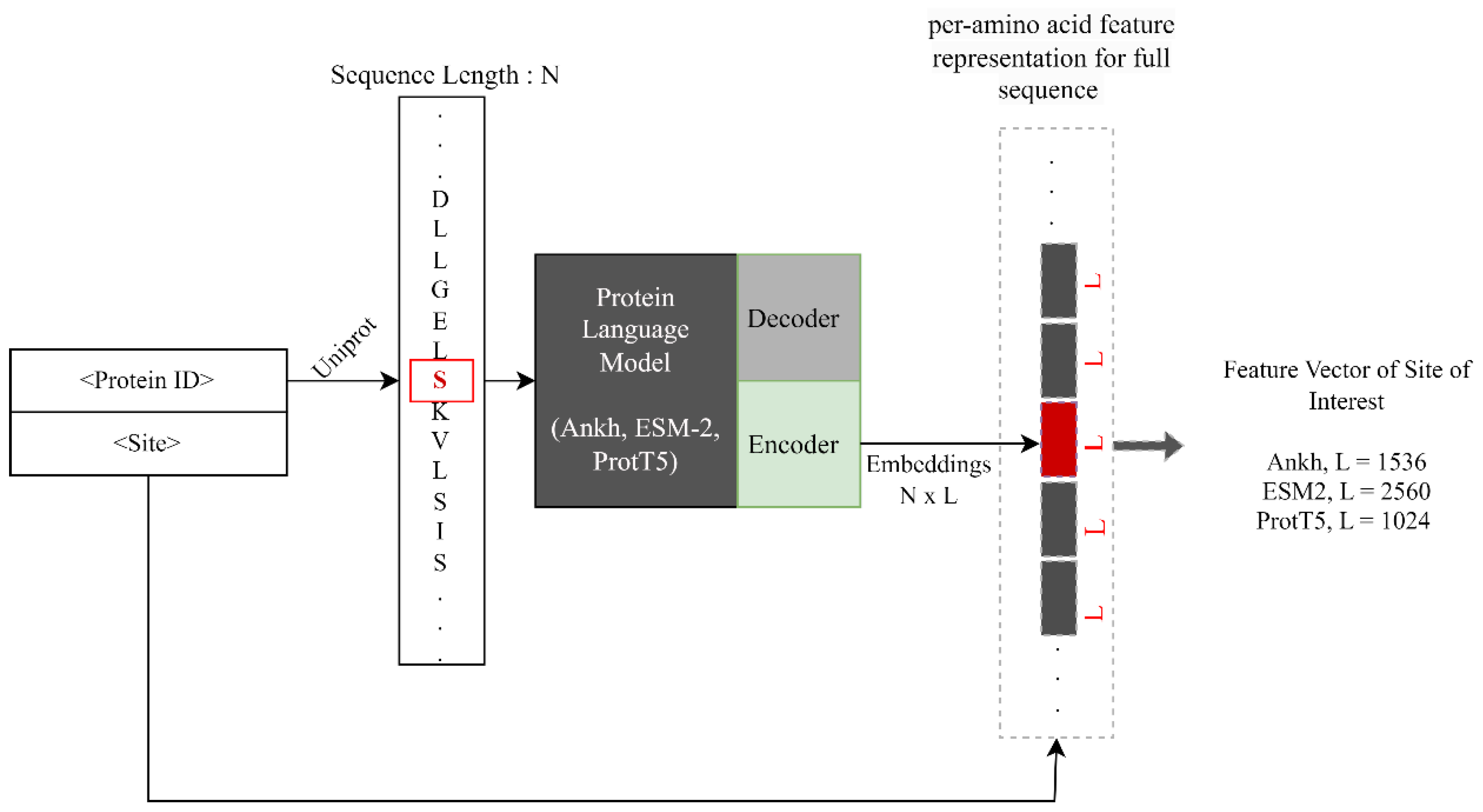
4.2. Sequence Encoding Using pLMs
4.2.1. Ankh
4.2.2. ESM-2
4.2.3. ProtT5
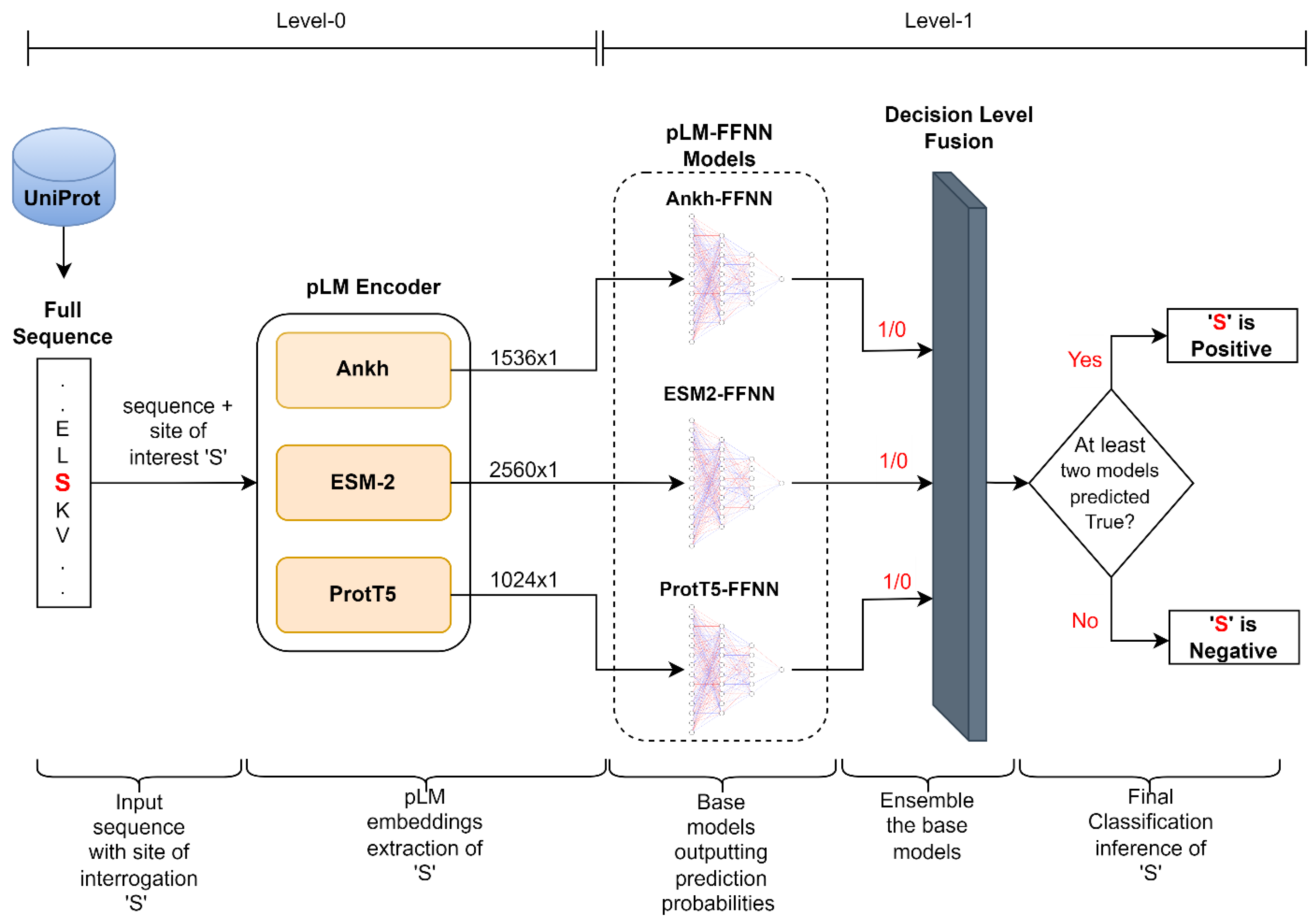
4.3. Model Architecture
4.4. Model Training and Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiro, R.G. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R–56R. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Copeland, R.J. Glycomics hits the big time. Cell 2010, 143, 672–676. [Google Scholar] [CrossRef]
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Holt, G.D.; Hart, G.W. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J. Biol. Chem. 1986, 261, 8049–8057. [Google Scholar] [CrossRef]
- Thompson, J.W.; Griffin, M.E.; Hsieh-Wilson, L.C. Methods for the detection, study, and dynamic profiling of O-GlcNAc glycosylation. Methods Enzymol. 2018, 598, 101–135. [Google Scholar] [CrossRef]
- Chatham, J.C.; Zhang, J.; Wende, A.R. Role of O-Linked N-Acetylglucosamine Protein Modification in Cellular (Patho)Physiology. Physiol. Rev. 2021, 101, 427–493. [Google Scholar] [CrossRef]
- Zachara, N.; Akimoto, Y.; Hart, G.W. The O-GlcNAc Modification. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Ma, J.; Wu, C.; Hart, G.W. Analytical and Biochemical Perspectives of Protein O-GlcNAcylation. Chem. Rev. 2021, 121, 1513–1581. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.C.; Chalkley, R.J. Methods for Enrichment and Assignment of N-Acetylglucosamine Modification Sites. Mol. Cell. Proteom. MCP 2021, 20, 100031. [Google Scholar] [CrossRef]
- Wang, J.; Torii, M.; Liu, H.; Hart, G.W.; Hu, Z.-Z. dbOGAP—An Integrated Bioinformatics Resource for Protein O-GlcNAcylation. BMC Bioinform. 2011, 12, 91. [Google Scholar] [CrossRef]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Olivier-Van Stichelen, S. The human O-GlcNAcome database and meta-analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.; Hou, C.; Wu, C. O-GlcNAcAtlas: A database of experimentally identified O-GlcNAc sites and proteins. Glycobiology 2021, 31, 719–723. [Google Scholar] [CrossRef]
- Olivier-Van Stichelen, S.; Malard, F.; Berendt, R.; Wulff-Fuentes, E.; Danner, L. Find out if your protein is O-GlcNAc modified: The O-GlcNAc database. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. In Pacific Symposium on Biocomputing 2002; World Scientific: Singapore, 2002; pp. 310–322. [Google Scholar]
- Abrahams, J.L.; Taherzadeh, G.; Jarvas, G.; Guttman, A.; Zhou, Y.; Campbell, M.P. Recent advances in glycoinformatic platforms for glycomics and glycoproteomics. Curr. Opin. Struct. Biol. 2020, 62, 56–69. [Google Scholar] [CrossRef]
- Bojar, D.; Lisacek, F. Glycoinformatics in the Artificial Intelligence Era. Chem. Rev. 2022, 122, 15971–15988. [Google Scholar] [CrossRef]
- Zhao, X.; Ning, Q.; Chai, H.; Ai, M.; Ma, Z. PGlcS: Prediction of protein O-GlcNAcylation sites with multiple features and analysis. J. Theor. Biol. 2015, 380, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.-J.; Huang, C.-H.; Bretaña, N.A.; Lu, C.-T.; Huang, K.-Y.; Weng, S.-L.; Lee, T.-Y. A two-layered machine learning method to identify protein O-GlcNAcylation sites with O-GlcNAc transferase substrate motifs. BMC Bioinform. 2015, 16, S10. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.-Z.; Liu, T.; Wang, Z.-P. O-GlcNAcPRED: A sensitive predictor to capture protein O-GlcNAcylation sites. Mol. Biosyst. 2013, 9, 2909–2913. [Google Scholar] [CrossRef]
- Jia, C.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinform. Oxf. Engl. 2018, 34, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Mohl, J.E.; Gerken, T.A.; Leung, M.-Y. ISOGlyP: De novo prediction of isoform-specific mucin-type O-glycosylation. Glycobiology 2020, 31, 168–172. [Google Scholar] [CrossRef]
- Elnaggar, A.; Essam, H.; Salah-Eldin, W.; Moustafa, W.; Elkerdawy, M.; Rochereau, C.; Rost, B. Ankh☥: Optimized protein language model unlocks general-purpose modelling. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef]
- Roshan Rao; Joshua Meier; Tom Sercu; Sergey Ovchinnikov; Alexander Rives Transformer protein language models are unsupervised structure learners. bioRxiv 2020. [CrossRef]
- Xiao, Y.; Qiu, J.; Li, Z.; Hsieh, C.-Y.; Tang, J. Modeling protein using large-scale pretrain language model. arXiv 2021, arXiv:2108.07435. [Google Scholar]
- Jozefowicz, R.; Vinyals, O.; Schuster, M.; Shazeer, N.; Wu, Y. Exploring the limits of language modeling. arXiv 2016, arXiv:1602.02410. [Google Scholar]
- Hou, X.; Wang, Y.; Bu, D.; Wang, Y.; Sun, S. EMNGly: Predicting N-linked glycosylation sites using the language models for feature extraction. Bioinformatics 2023, 39, btad650. [Google Scholar] [CrossRef]
- Pakhrin, S.C.; Pokharel, S.; Aoki-Kinoshita, K.F.; Beck, M.R.; Dam, T.K.; Caragea, D.; Kc, D.B. LMNglyPred: Prediction of human N-linked glycosylation sites using embeddings from a pre-trained protein language model. Glycobiology 2023, 33, 411–422. [Google Scholar] [CrossRef]
- Alkuhlani, A.; Gad, W.; Roushdy, M.; Voskoglou, M.G.; Salem, A.-b.M. PTG-PLM: Predicting Post-Translational Glycosylation and Glycation Sites Using Protein Language Models and Deep Learning. Axioms 2022, 11, 469. [Google Scholar] [CrossRef]
- Pratyush, P.; Pokharel, S.; Saigo, H.; Kc, D.B. pLMSNOSite: An ensemble-based approach for predicting protein S-nitrosylation sites by integrating supervised word embedding and embedding from pre-trained protein language model. BMC Bioinform. 2023, 24, 41. [Google Scholar] [CrossRef]
- Unsal, S.; Atas, H.; Albayrak, M.; Turhan, K.; Acar, A.C.; Doğan, T. Learning functional properties of proteins with language models. Nat. Mach. Intell. 2022, 4, 227–245. [Google Scholar] [CrossRef]
- Marquet, C.; Heinzinger, M.; Olenyi, T.; Dallago, C.; Erckert, K.; Bernhofer, M.; Nechaev, D.; Rost, B. Embeddings from protein language models predict conservation and variant effects. Hum. Genet. 2022, 141, 1629–1647. [Google Scholar] [CrossRef]
- Jha, K.; Karmakar, S.; Saha, S. Graph-BERT and language model-based framework for protein–protein interaction identification. Sci. Rep. 2023, 13, 5663. [Google Scholar] [CrossRef]
- Villegas-Morcillo, A.; Gomez, A.M.; Sanchez, V. An analysis of protein language model embeddings for fold prediction. Brief. Bioinform. 2022, 23, bbac142. [Google Scholar] [CrossRef]
- Nallapareddy, V.; Bordin, N.; Sillitoe, I.; Heinzinger, M.; Littmann, M.; Waman, V.P.; Sen, N.; Rost, B.; Orengo, C. CATHe: Detection of remote homologues for CATH superfamilies using embeddings from protein language models. Bioinformatics 2023, 39, btad029. [Google Scholar] [CrossRef]
- Tazhitdinova, R.; Timoshenko, A.V. The Emerging Role of Galectins and O-GlcNAc Homeostasis in Processes of Cellular Differentiation. Cells 2020, 9, 1792. [Google Scholar] [CrossRef]
- Hu, G.; Katuwawala, A.; Wang, K.; Wu, Z.; Ghadermarzi, S.; Gao, J.; Kurgan, L. flDPnn: Accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat. Commun. 2021, 12, 4438. [Google Scholar] [CrossRef]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar]
- Villegas-Morcillo, A.; Makrodimitris, S.; van Ham, R.C.H.J.; Gomez, A.M.; Sanchez, V.; Reinders, M.J.T. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics 2021, 37, 162–170. [Google Scholar] [CrossRef]
- van den Bent, I.; Makrodimitris, S.; Reinders, M. The Power of Universal Contextualized Protein Embeddings in Cross-species Protein Function Prediction. Evol. Bioinform. Online 2021, 17, 11769343211062608. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 2020, 21, 5485–5551. [Google Scholar]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Mean Accuracy | Mean Sensitivity | Mean Specificity | Mean MCC | Mean ROC-AUC |
|---|---|---|---|---|---|
| Ankh-FFNN | 0.7141 (0.0078) | 0.7236 (0.0267) | 0.7047 (0.0287) | 0.4289 (0.0152) | 0.7915 (0.0104) |
| ESM2-FFNN | 0.7026 (0.0130) | 0.7497 (0.0480) | 0.6556 (0.0615) | 0.4092 (0.0235) | 0.7530 (0.0243) |
| ProtT5-FFNN | 0.7152 (0.0068) | 0.7352 (0.0073) | 0.6952 (0.0149) | 0.4308 (0.0134) | 0.7872 (0.0105) |
| Model | Feature Type | Accuracy | Sensitivity | Specificity | MCC | ROC-AUC |
|---|---|---|---|---|---|---|
| Individual Models | Ankh | 0.7126 | 0.7122 | 0.7325 | 0.1329 | 0.7962 |
| ESM-2 | 0.7059 | 0.7071 | 0.6457 | 0.1050 | 0.7438 | |
| ProtT5 | 0.6999 | 0.6981 | 0.8263 | 0.1334 | 0.8348 | |
| Score-Level Fusion | Ankh + ESM-2 + ProtT5 | 0.7495 | 0.7502 | 0.7141 | 0.1447 | 0.8116 |
| Ankh + ESM-2 | 0.7394 | 0.7397 | 0.7229 | 0.1423 | 0.8111 | |
| Ankh + ESM-2 | 0.7358 | 0.7364 | 0.7038 | 0.1350 | 0.7954 | |
| ESM-2 + ProtT5 | 0.7396 | 0.7402 | 0.7078 | 0.1379 | 0.7985 | |
| Decision-Level Fusion (LM-OGlcNAc-Site) | Ankh + ESM-2 + ProtT5 | 0.8590 | 0.8648 | 0.5613 | 0.1659 | 0.8116 |
| Model | Feature Type | Accuracy | Sensitivity | Specificity | MCC | ROC-AUC |
|---|---|---|---|---|---|---|
| Individual Model | Ankh | 0.6767 | 0.7899 | 0.6751 | 0.1161 | 0.8077 |
| ESM-2 | 0.5833 | 0.8123 | 0.5800 | 0.1270 | 0.7717 | |
| ProtT5 | 0.6765 | 0.8347 | 0.6742 | 0.1270 | 0.8348 | |
| Score-Level Fusion | Ankh + ESM-2 + ProtT5 | 0.6634 | 0.8263 | 0.6611 | 0.1205 | 0.8320 |
| Ankh + ESM-2 | 0.6871 | 0.8179 | 0.6852 | 0.1266 | 0.8350 | |
| Ankh + ESM-2 | 0.6350 | 0.8095 | 0.6326 | 0.1074 | 0.8117 | |
| ESM-2 + ProtT5 | 0.6354 | 0.8571 | 0.6323 | 0.1189 | 0.8258 | |
| Decision-Level Fusion (LM-OGlcNAc-Site) | Ankh + ESM-2 + ProtT5 | 0.7836 | 0.7115 | 0.7846 | 0.1403 | 0.8320 |
| Model | Feature Type | Accuracy | Sensitivity | Specificity | MCC | ROC-AUC |
|---|---|---|---|---|---|---|
| Individual Model | Ankh | 0.7548 | 0.6162 | 0.7568 | 0.1015 | 0.7468 |
| ESM-2 | 0.6834 | 0.7199 | 0.6829 | 0.1014 | 0.7561 | |
| ProtT5 | 0.7263 | 0.7086 | 0.7265 | 0.1140 | 0.7707 | |
| Score-Level Fusion | Ankh + ESM-2 + ProtT5 | 0.7367 | 0.7148 | 0.7371 | 0.1189 | 0.7892 |
| Ankh + ESM-2 | 0.7232 | 0.7030 | 0.7235 | 0.1114 | 0.7812 | |
| Ankh + ESM-2 | 0.7314 | 0.6946 | 0.7319 | 0.1124 | 0.7820 | |
| ESM-2 + ProtT5 | 0.7314 | 0.7254 | 0.7275 | 0.1188 | 0.7803 | |
| Decision-Level Fusion (LM-OGlcNAc-Site) | Ankh + ESM-2 + ProtT5 | 0.8695 | 0.5042 | 0.8747 | 0.1322 | 0.7892 |
| O-GlcNAcPRED-II [20] | 0.7239 | 0.6712 | 0.7246 | 0.1012 | 0.7433 | |
| Galectin | NCBI Reference Sequence | OGTSite | YinOYang | LM-OGlcNAc-Site |
|---|---|---|---|---|
| Galectin-1 | NP_002296.1 | S84 | S84 | T71, S84 |
| Galectin-2 | NP_006489.1 | S23, S51, S80, T85, S122 | no predictions | T21, S80, T85 |
| Galectin-3 | NP_002297.2 | S84, T133 | S84, S91, S92, T98, T104, T243 | S6, S12, S14, S40, S84, S91, S92, S96, T98, T104 |
| Galectin-4 | NP_006140.1 | S40 | S58, T217, T317 | S86, S92, T217, S258 |
| Galectin-7 | NP_002298.1 | S9 | S2, S8, S9, S45, T57, T58 | S2, T18, S31, S69 |
| Galectin-8 | NP_006490.3 | T22, S152, T160, T201, T211 | T160, S178, S358 | S4, T160, S171, S178, T180, S192, T198, T201, S203, T207, T211, T215 |
| Galectin-9 | NP_033665.1 | S18, T32, T152, S165, T193, S202 | S4, S6, S12, S18, S139, T152, S160, T161, S165, T193, T195, T351, T355 | S4, S6, T152, S160, S161, S165, T193, T195 |
| Galectin-10 | NP_001819.2 | T9 | T9, T16 | S13 |
| Galectin-12 | NP_001136007.2 | T81, T82, S143, S192, S221, T232, S305 | T82, S192, T232, S315 | S2, S221 |
| Galectin-13 | NP_037400.1 | S13, S119, S127, T133 | S2, S3, S13, S119, S127, T133 | S2, S13 |
| Galectin-14 | NP_064514.1 | T9, S13 | S2, S3, T9, S13, S138 | S2, S13, S138 |
| Galectin-16 | NP_001177370.2 | S13, S119 | S13, S119 | S2, S13 |
| No. of Proteins | Positive Sites | Negative Sites | ||
|---|---|---|---|---|
| Train | 4826 | 12,644 | Before Under-Sampling | 662,081 |
| After Under-Sampling | 12,644 | |||
| Test | 529 | 1256 | 64,927 | |
| pLM | Dataset | Architecture | Developer | Number of Parameters (Billions) | Embedding Dimension Per Residue (L × 1) |
|---|---|---|---|---|---|
| Ankh [22] | Uniref50 [44] | ConvBERT | Rostlab | 1.2B | 1536 × 1 |
| ESM-2 (esm2_t36_3B_UR50D) [23] | RoBERTa | Meta | 3B | 2560 × 1 | |
| ProtT5 (ProtT5-XL-U50) [24] | T5 | Rostlab | 3B | 1024 × 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokharel, S.; Pratyush, P.; Ismail, H.D.; Ma, J.; KC, D.B. Integrating Embeddings from Multiple Protein Language Models to Improve Protein O-GlcNAc Site Prediction. Int. J. Mol. Sci. 2023, 24, 16000. https://doi.org/10.3390/ijms242116000
Pokharel S, Pratyush P, Ismail HD, Ma J, KC DB. Integrating Embeddings from Multiple Protein Language Models to Improve Protein O-GlcNAc Site Prediction. International Journal of Molecular Sciences. 2023; 24(21):16000. https://doi.org/10.3390/ijms242116000
Chicago/Turabian StylePokharel, Suresh, Pawel Pratyush, Hamid D. Ismail, Junfeng Ma, and Dukka B. KC. 2023. "Integrating Embeddings from Multiple Protein Language Models to Improve Protein O-GlcNAc Site Prediction" International Journal of Molecular Sciences 24, no. 21: 16000. https://doi.org/10.3390/ijms242116000
APA StylePokharel, S., Pratyush, P., Ismail, H. D., Ma, J., & KC, D. B. (2023). Integrating Embeddings from Multiple Protein Language Models to Improve Protein O-GlcNAc Site Prediction. International Journal of Molecular Sciences, 24(21), 16000. https://doi.org/10.3390/ijms242116000