
Identification of Hub Genes in Idiopathic Pulmonary Fibrosis and Their Association with Lung Cancer by Bioinformatics Analysis

 ,
,  , , and
, , and
Abstract
:Highlights
- Identification of 1888 differentially expressed genes (DEGs) related to idiopathic pulmonary fibrosis (IPF), including 1105 upregulated and 783 downregulated genes.
- Discovery of 10 hub genes with high connectivity that may play a crucial role in the pathogenesis of IPF, with implications for potential diagnostic biomarkers and therapeutic targets.
- The study sheds light on the genetic landscape of IPF, uncovering potential key players in its development and progression.
- These identified hub genes have relevance beyond IPF, being expressed in lung cancer and associated with various stages of cancer progression, suggesting a link between IPF and lung cancer that could pave the way for improved diagnostics and therapies.
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Selection and Inclusion Criteria for Gene Expression Microarray Data
2.3. Identification of DEGs
2.4. GO Enrichment and KEGG Pathway Enrichment Analyses
2.5. PPI Network Analysis
2.6. Validation of the Hub Genes
2.7. Analysis for miRNA Target Gene Prediction and miRNA-mRNA Network Construction
2.8. Analysis of Hub Gene Expression in Lung Cancer
3. Results
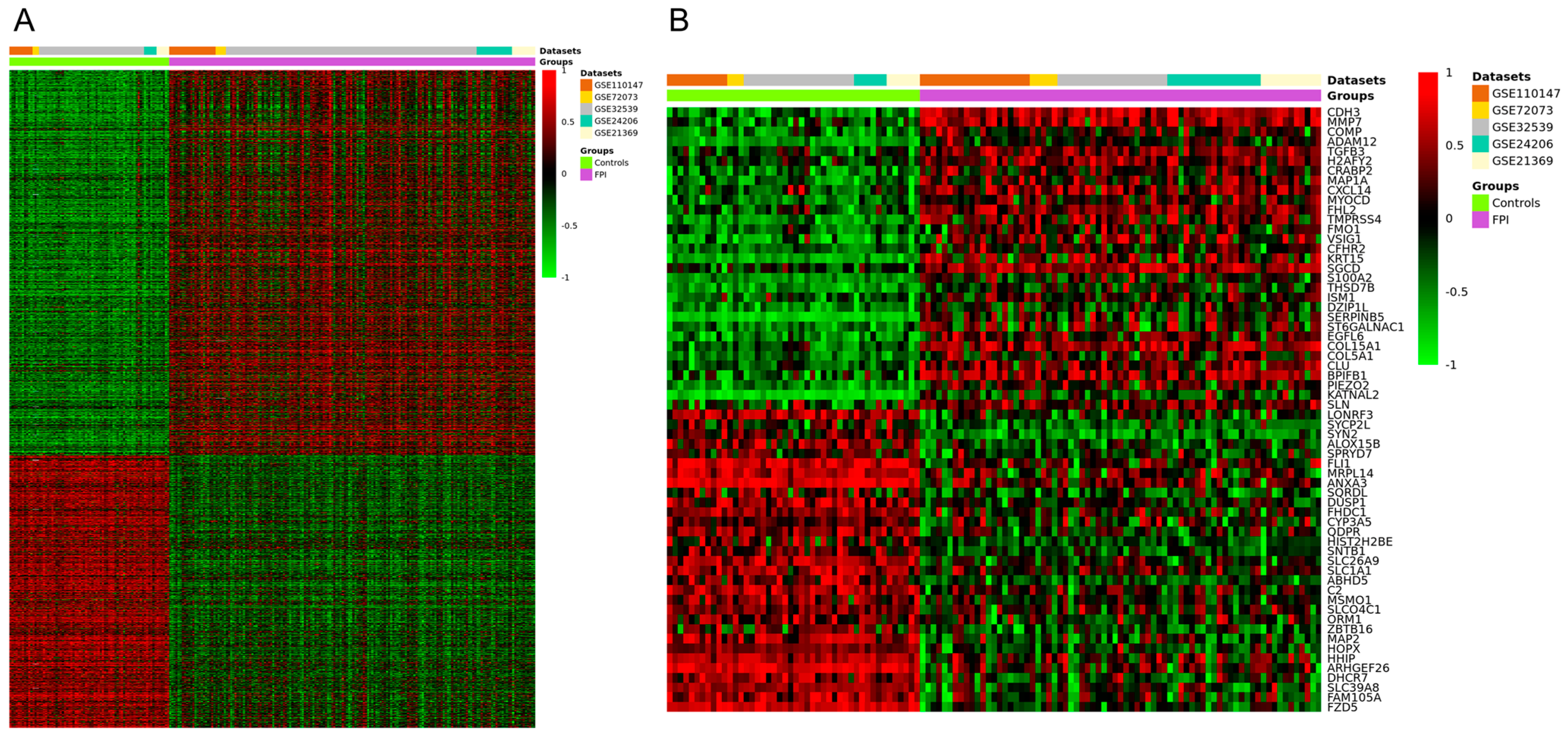
3.1. Processing of Microarray Datasets and Identification of DEGs in IPF
3.2. GO Enrichment and KEGG Pathway Analyses for DEGs in IPF
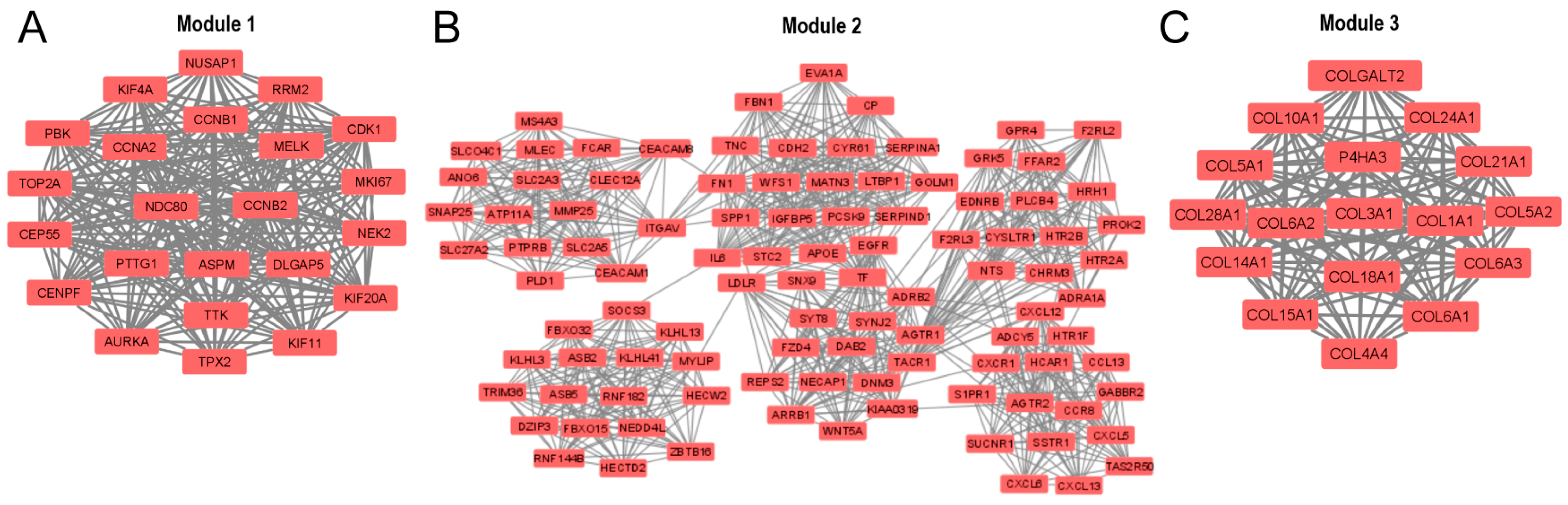
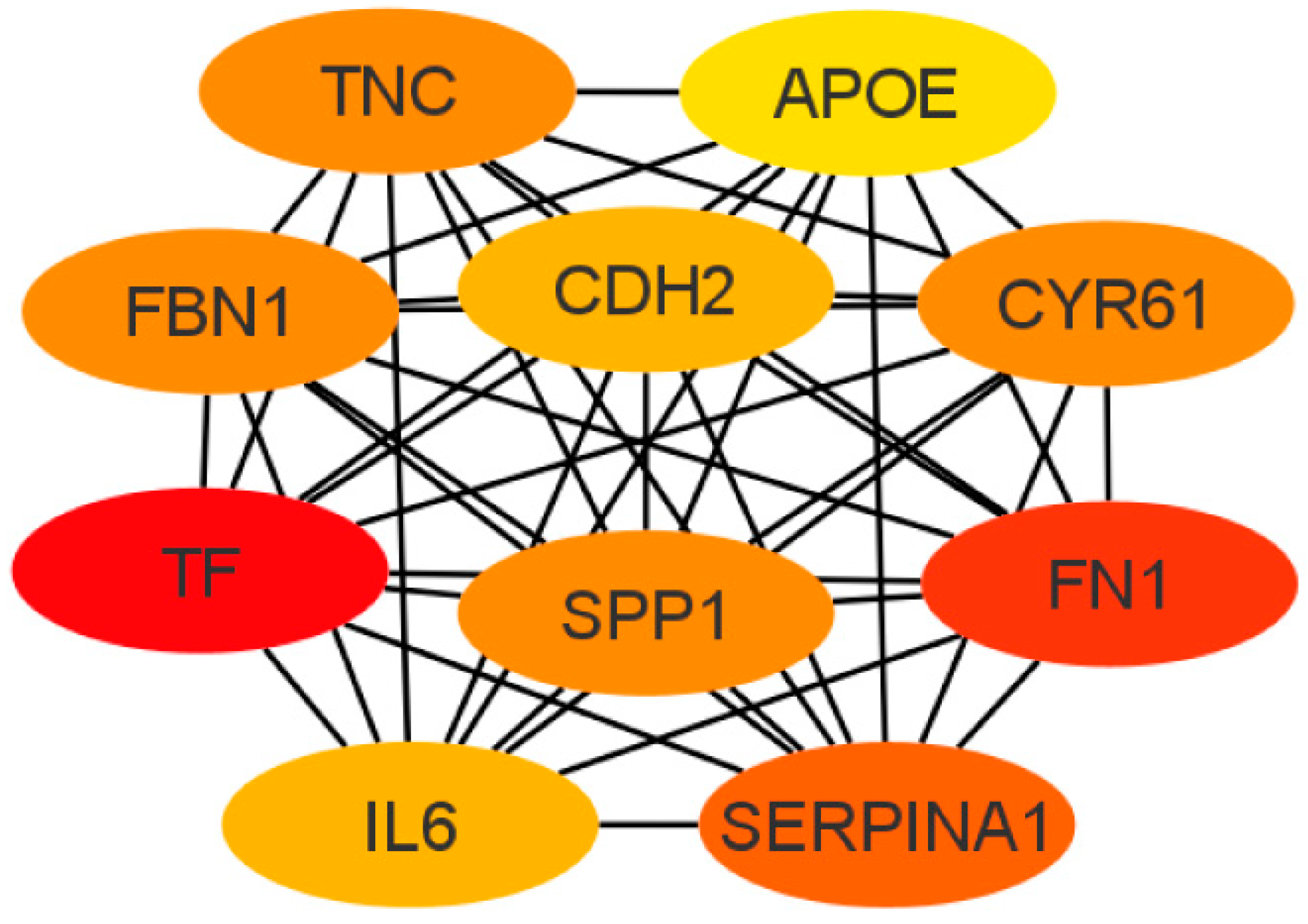
3.3. PPI Network and Identification of Hub Genes
3.4. GO and KEGG Pathway Enrichment Analysis for the DEGs Present in the Three Main Modules of the PPI Network
3.5. GO Enrichment and KEGG Pathway Analyses for the Top 10 Hub Genes in the PPI Network
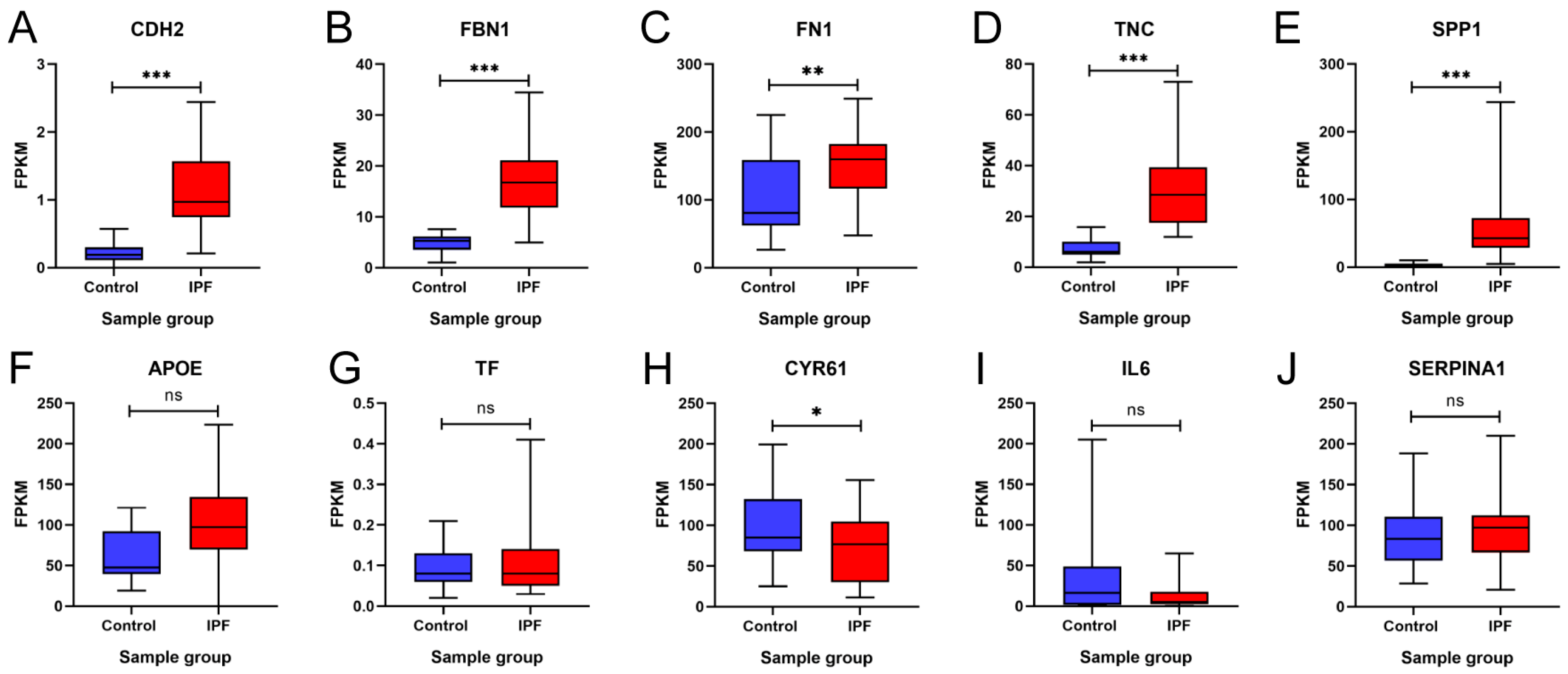
3.6. In Silico Validation of Hub Genes
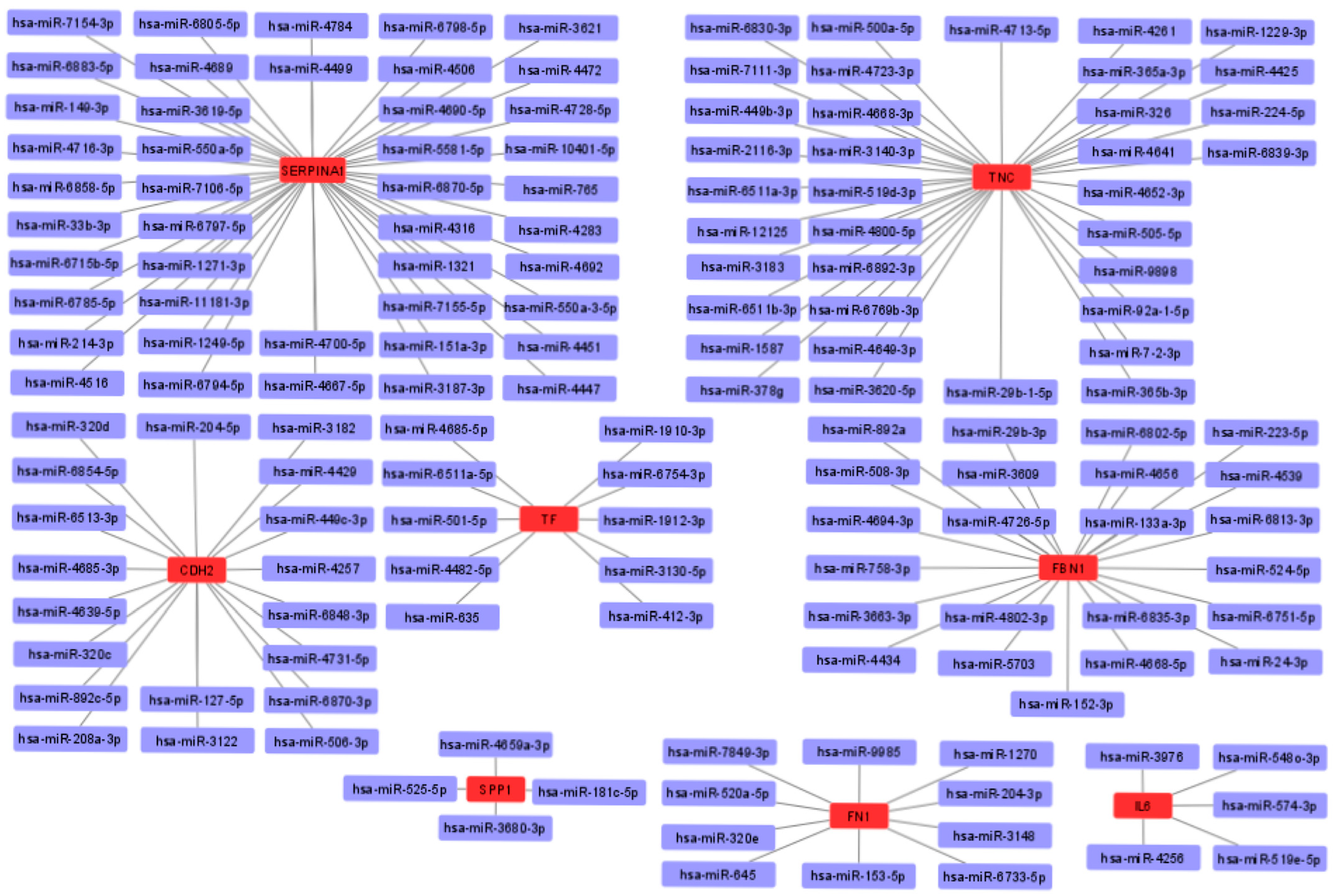
3.7. miRNA-mRNA Interaction Prediction and Network
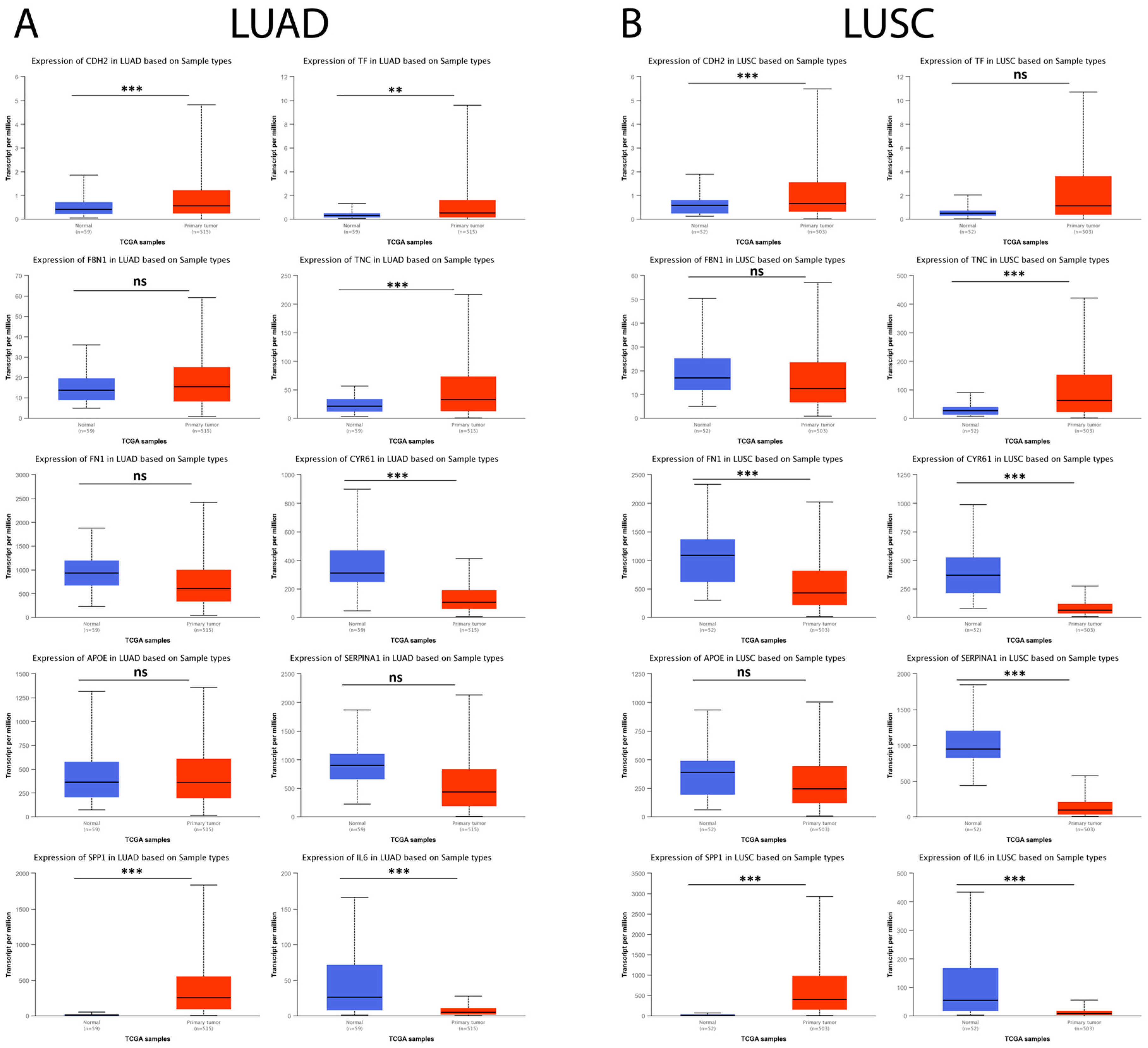
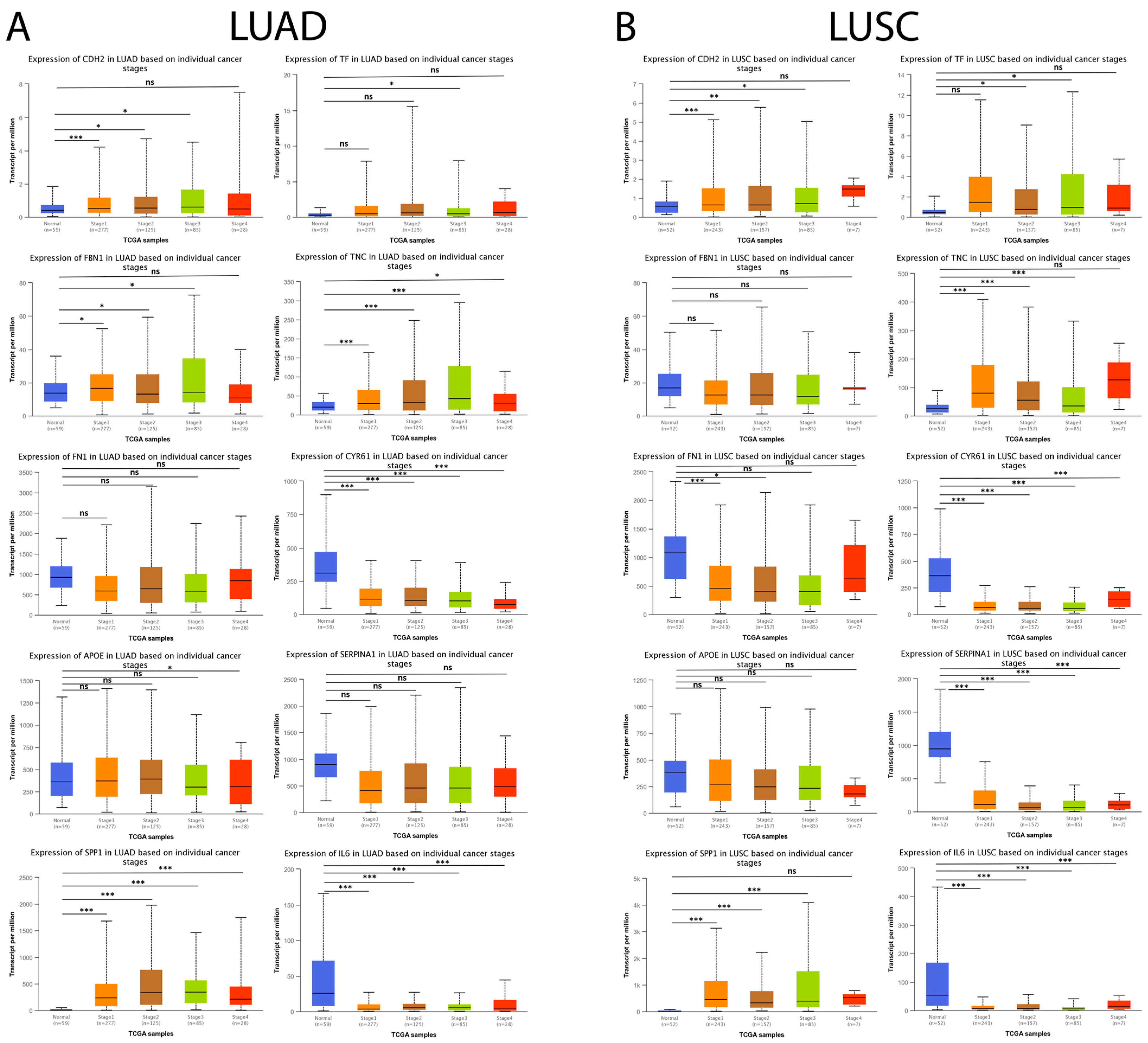
3.8. Hub Genes in IPF Are Expressed in Lung Cancer and Are Associated with Cancer Progression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Hu, Y. TGF-β1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- du Bois, R.M. An earlier and more confident diagnosis of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2012, 21, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.W.; Tan, K.; Yang, W.; Zhao, H.; Wang, G.Q. Discharge may not be the end of treatment: Pay attention to pulmonary fibrosis caused by severe COVID-19. J. Med. Virol. 2021, 93, 1378–1386. [Google Scholar] [CrossRef]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Casa, G.D.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet Respir. Med. 2020, 8, 750–752. [Google Scholar] [CrossRef]
- Nakamura, Y.; Suda, T. Idiopathic Pulmonary Fibrosis: Diagnosis and Clinical Manifestations. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Grodkiewicz, M.; Koziel, P.; Chmielewska, I.; Korbel, M.A.; Milanowski, J. Small Cell Lung Cancer in the Course of Idiopathic Pulmonary Fibrosis—Case Report and Literature Review. Curr. Oncol. 2022, 29, 5077–5083. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Kato, E.; Takayanagi, N.; Takaku, Y.; Kagiyama, N.; Kanauchi, T.; Ishiguro, T.; Sugita, Y. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2018, 4, 00111–2016. [Google Scholar] [CrossRef]
- Fan, L.; Yu, X.; Huang, Z.; Zheng, S.; Zhou, Y.; Lv, H.; Zeng, Y.; Xu, J.F.; Zhu, X.; Yi, X. Analysis of Microarray-Identified Genes and MicroRNAs Associated with Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2017, 2017, 1804240. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Wang, B. Identification of transcriptomic markers for developing idiopathic pulmonary fibrosis: An integrative analysis of gene expression profiles. Int. J. Clin. Exp. Pathol. 2020, 13, 1698–1706. [Google Scholar]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Meltzer, E.B.; Barry, W.T.; D’Amico, T.A.; Davis, R.D.; Lin, S.S.; Onaitis, M.W.; Morrison, L.D.; Sporn, T.A.; Steele, M.P.; Noble, P.W. Bayesian probit regression model for the diagnosis of pulmonary fibrosis: Proof-of-principle. BMC Med. Genom. 2011, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Gelinas, R.; Wang, K.; Etheridge, A.; Piper, M.G.; Batte, K.; Dakhallah, D.; Price, J.; Bornman, D.; Zhang, S.; et al. Systems biology of interstitial lung diseases: Integration of mRNA and microRNA expression changes. BMC Med. Genom. 2011, 4, 8. [Google Scholar] [CrossRef]
- Cecchini, M.J.; Hosein, K.; Howlett, C.J.; Joseph, M.; Mura, M. Comprehensive gene expression profiling identifies distinct and overlapping transcriptional profiles in non-specific interstitial pneumonia and idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 153. [Google Scholar] [CrossRef]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Coldren, C.D.; Leach, S.M.; Seibold, M.A.; Murphy, E.; Lin, J.; Rosen, R.; Neidermyer, A.J.; McKean, D.F.; Groshong, S.D.; et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 2013, 68, 1114–1121. [Google Scholar] [CrossRef]
- Xu, Z.; Mo, L.; Feng, X.; Huang, M.; Li, L. Using bioinformatics approach identifies key genes and pathways in idiopathic pulmonary fibrosis. Medicine 2020, 99, e22099. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, Q.; Ou-Yang, W.; Zhang, M. Integrative analyses of genes associated with idiopathic pulmonary fibrosis. J. Cell. Biochem. 2018, 120, 8648–8660. [Google Scholar] [CrossRef]
- Toro-Domínguez, D.; Martorell-Marugán, J.; López-Domínguez, R.; García-Moreno, A.; González-Rumayor, V.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. ImaGEO: Integrative gene expression meta-analysis from GEO database. Bioinformatics 2019, 35, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Z.; Hu, Y.F.; Zhang, Y.; Wei, S.Y.; Yang, B.L.; Xu, Y.P.; Rong, Z.L.; Wang, D.; Yang, B. FibROAD: A manually curated resource for multi-omics level evidence integration of fibrosis research. Database 2022, 2022, baac015. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Lagares, D.; Busnadiego, O.; Garcia-Fernandez, R.A.; Kapoor, M.; Liu, S.; Carter, D.E.; Abraham, D.; Shi-Wen, X.; Carreira, P.; Fontaine, B.A.; et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012, 64, 1653–1664. [Google Scholar] [CrossRef]
- Zhao, X.K.; Cheng, Y.; Liang Cheng, M.; Yu, L.; Mu, M.; Li, H.; Liu, Y.; Zhang, B.; Yao, Y.; Guo, H.; et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Sci. Rep. 2016, 6, 19276. [Google Scholar] [CrossRef]
- Estany, S.; Vicens-Zygmunt, V.; Llatjós, R.; Montes, A.; Penín, R.; Escobar, I.; Xaubet, A.; Santos, S.; Manresa, F.; Dorca, J.; et al. Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFβ1. BMC Pulm. Med. 2014, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Otoupalova, E.; Hough, K.P.; Locy, M.L.; Bernard, K.; Deshane, J.S.; Sanderson, R.D.; Mobley, J.A.; Thannickal, V.J. Fibronectin on the Surface of Extracellular Vesicles Mediates Fibroblast Invasion. Am. J. Respir. Cell Mol. Biol. 2019, 60, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Qiu, L.; Zeng, C.; Fang, Z.; Chen, S.; Song, X.; Song, H.; Zhang, G. Bioinformatic analysis of differentially expressed genes and pathways in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2021, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Gong, G.; Wu, W.; Li, N.; Li, Z.; Chen, S.; Li, P.; Chen, T.; Zhao, H.; Hu, C.; et al. A novel prognostic signature for idiopathic pulmonary fibrosis based on five-immune-related genes. Ann. Transl. Med. 2021, 9, 1570. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Xia, Q.; Jiang, W.; Zhang, H.; Amiri-Ardekani, E.; Hua, H.; Cheng, Y. Machine learning-based prediction of candidate gene biomarkers correlated with immune infiltration in patients with idiopathic pulmonary fibrosis. Front. Med. 2023, 10, 1001813. [Google Scholar] [CrossRef]
- Kang, J.; Yeo, H.J.; Kim, Y.H.; Cho, W.H. Molecular differences between stable idiopathic pulmonary fibrosis and its acute exacerbation. Front. Biosci. 2021, 26, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Yang, Z.; Zhang, W.; Liu, S.; Zhao, Q.; Liu, T.; Chen, L.; Li, L.; Wang, Y.; Shao, R. Identification of diagnostic gene biomarkers related to immune infiltration in patients with idiopathic pulmonary fibrosis based on bioinformatics strategies. Front. Med. 2022, 9, 959010. [Google Scholar] [CrossRef]
- Fan, G.; Liu, J.; Wu, Z.; Li, C.; Zhang, Y. Development and validation of the prognostic model based on autophagy-associated genes in idiopathic pulmonary fibrosis. Front. Immunol. 2022, 13, 1049361. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef] [PubMed]
- Carey, W.A.; Taylor, G.D.; Dean, W.B.; Bristow, J.D. Tenascin-C deficiency attenuates TGF-ß-mediated fibrosis following murine lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L785–L793. [Google Scholar] [CrossRef]
- Vij, R.; Noth, I. Peripheral blood biomarkers in idiopathic pulmonary fibrosis. Transl. Res. J. Lab. Clin. Med. 2012, 159, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef]
- Gui, X.; Qiu, X.; Xie, M.; Tian, Y.; Min, C.; Huang, M.; Hongyan, W.; Chen, T.; Zhang, X.; Chen, J.; et al. Prognostic Value of Serum Osteopontin in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. BioMed. Res. Int. 2020, 2020, 3424208. [Google Scholar] [CrossRef]
- Berman, J.S.; Serlin, D.; Li, X.; Whitley, G.; Hayes, J.; Rishikof, D.C.; Ricupero, D.A.; Liaw, L.; Goetschkes, M.; O’Regan, A.W. Altered bleomycin-induced lung fibrosis in osteopontin-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L1311–L1318. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, F.; Takahashi, K.; Okazaki, T.; Maeda, K.; Ienaga, H.; Maeda, M.; Kon, S.; Uede, T.; Fukuchi, Y. Role of osteopontin in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2001, 24, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Hatipoglu, O.F.; Uctepe, E.; Opoku, G.; Wake, H.; Ikemura, K.; Ohtsuki, T.; Inagaki, J.; Gunduz, M.; Gunduz, E.; Watanabe, S.; et al. Osteopontin silencing attenuates bleomycin-induced murine pulmonary fibrosis by regulating epithelial-mesenchymal transition. Biomed. Pharmacother. 2021, 139, 111633. [Google Scholar] [CrossRef]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005041. [Google Scholar] [CrossRef] [PubMed]
- Muro, A.F.; Moretti, F.A.; Moore, B.B.; Yan, M.; Atrasz, R.G.; Wilke, C.A.; Flaherty, K.R.; Martinez, F.J.; Tsui, J.L.; Sheppard, D.; et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 638–645. [Google Scholar] [CrossRef]
- Andugulapati, S.B.; Gourishetti, K.; Tirunavalli, S.K.; Shaikh, T.B.; Sistla, R. Biochanin—A ameliorates pulmonary fibrosis by suppressing the TGF-β mediated EMT, myofibroblasts differentiation and collagen deposition in in vitro and in vivo systems. Phytomedicine 2020, 78, 153298. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Machahua-Huamani, C.; Vicens-Zygmunt, V.; Llatjós, R.; Escobar, I.; Sala-Llinas, E.; Luburich-Hernaiz, P.; Dorca, J.; Montes-Worboys, A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018, 18, 63. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef]
- Zhang, R.M.; Zeyer, K.A.; Odenthal, N.; Zhang, Y.; Reinhardt, D.P. The fibrillin-1 RGD motif posttranscriptionally regulates ERK1/2 signaling and fibroblast proliferation via miR-1208. Faseb J. 2021, 35, e21598. [Google Scholar] [CrossRef]
- Xu, H.; Zaidi, M.; Struve, J.; Jones, D.W.; Krolikowski, J.G.; Nandedkar, S.; Lohr, N.L.; Gadicherla, A.; Pagel, P.S.; Csuka, M.E.; et al. Abnormal fibrillin-1 expression and chronic oxidative stress mediate endothelial mesenchymal transition in a murine model of systemic sclerosis. Am. J. Physiol. Cell Physiol. 2011, 300, C550–C556. [Google Scholar] [CrossRef] [PubMed]
- Lorena, D.; Darby, I.A.; Reinhardt, D.P.; Sapin, V.; Rosenbaum, J.; Desmoulière, A. Fibrillin-1 expression in normal and fibrotic rat liver and in cultured hepatic fibroblastic cells: Modulation by mechanical stress and role in cell adhesion. Lab. Investig. 2004, 84, 203–212. [Google Scholar] [CrossRef]
- Li, L.; Liao, J.; Yuan, Q.; Hong, X.; Li, J.; Peng, Y.; He, M.; Zhu, H.; Zhu, M.; Hou, F.F.; et al. Fibrillin-1-enriched microenvironment drives endothelial injury and vascular rarefaction in chronic kidney disease. Sci. Adv. 2021, 7, eabc7170. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Apoprotein E and Reverse Cholesterol Transport. Int. J. Mol. Sci. 2018, 19, 3479. [Google Scholar] [CrossRef]
- Yao, X.; Gordon, E.M.; Figueroa, D.M.; Barochia, A.V.; Levine, S.J. Emerging Roles of Apolipoprotein E and Apolipoprotein A-I in the Pathogenesis and Treatment of Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 55, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Madenspacher, J.H.; Azzam, K.M.; Gong, W.; Gowdy, K.M.; Vitek, M.P.; Laskowitz, D.T.; Remaley, A.T.; Wang, J.M.; Fessler, M.B. Apolipoproteins and apolipoprotein mimetic peptides modulate phagocyte trafficking through chemotactic activity. J. Biol. Chem. 2012, 287, 43730–43740. [Google Scholar] [CrossRef]
- Cui, H.; Jiang, D.; Banerjee, S.; Xie, N.; Kulkarni, T.; Liu, R.M.; Duncan, S.R.; Liu, G. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. J. Clin. Investig. 2020, 5, e134539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Cai, J.; Jiang, C. CDH2 expression is of prognostic significance in glioma and predicts the efficacy of temozolomide therapy in patients with glioblastoma. Oncol. Lett. 2018, 15, 7415–7422. [Google Scholar] [CrossRef]
- Cao, Z.-Q.; Wang, Z.; Leng, P. Aberrant N-cadherin expression in cancer. Biomed. Pharmacother. 2019, 118, 109320. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wen, L.; Shi, Q.F.; Gao, F.; Huang, B.; Meng, J.; Hu, C.P.; Wang, C.M. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial-Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef]
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhang, Y.; Zhang, Y.; Wang, X.; Xia, Y. Pterostilbene alleviates pulmonary fibrosis by regulating ASIC2. Chin. Med. 2021, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Head, J.F. Cancer: Tumor Iron Metabolism, Mitochondrial Dysfunction and Tumor Immunosuppression; “A Tight Partnership—Was Warburg Correct? ” J. Cancer Ther. 2012, 3, 34. [Google Scholar] [CrossRef]
- Ogger, P.P.; Byrne, A.J. Lung fibrosis enters the iron age(†). J. Pathol. 2020, 252, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.K.; Kim, R.Y.; Karim, R.; Mayall, J.R.; Martin, K.L.; Shahandeh, A.; Abbasian, F.; Starkey, M.R.; Loustaud-Ratti, V.; Johnstone, D.; et al. Role of iron in the pathogenesis of respiratory disease. Int. J. Biochem. Cell Biol. 2017, 88, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Puxeddu, E.; Comandini, A.; Cavalli, F.; Pezzuto, G.; D’Ambrosio, C.; Senis, L.; Paci, M.; Curradi, G.; Sergiacomi, G.L.; Saltini, C. Iron laden macrophages in idiopathic pulmonary fibrosis: The telltale of occult alveolar hemorrhage? Pulm. Pharmacol. Ther. 2014, 28, 35–40. [Google Scholar] [CrossRef]
- Ghio, A.J.; Carter, J.D.; Richards, J.H.; Richer, L.D.; Grissom, C.K.; Elstad, M.R. Iron and iron-related proteins in the lower respiratory tract of patients with acute respiratory distress syndrome. Crit. Care Med. 2003, 31, 395–400. [Google Scholar] [CrossRef]
- Matamala, N.; Lara, B.; Gómez-Mariano, G.; Martínez, S.; Vázquez-Domínguez, I.; Otero-Sobrino, Á.; Muñoz-Callejas, A.; Sánchez, E.; Esquinas, C.; Bustamante, A.; et al. miR-320c Regulates SERPINA1 Expression and Is Induced in Patients With Pulmonary Disease. Arch. Bronconeumol. 2020, 57, 457–463. [Google Scholar] [CrossRef]
- Tubío-Pérez, R.A.; Torres-Durán, M.; Fernández-Villar, A.; Ruano-Raviña, A. Alpha-1 antitrypsin deficiency and risk of lung cancer: A systematic review. Transl. Oncol. 2021, 14, 100914. [Google Scholar] [CrossRef] [PubMed]
- Afonso, M.; Silva, C.; Pinho, I.; Vale, A.; Fernandes, A. A rare mutation on alpha-1 antitrypsin deficit and lung fibrosis: Case report. Sarcoidosis Vasc. Diffus. Lung Dis. 2020, 37, e2020019. [Google Scholar] [CrossRef]
- Zhu, Y.; Almuntashiri, S.; Han, Y.; Wang, X.; Somanath, P.R.; Zhang, D. The Roles of CCN1/CYR61 in Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 7810. [Google Scholar] [CrossRef]
- Grazioli, S.; Gil, S.; An, D.; Kajikawa, O.; Farnand, A.W.; Hanson, J.F.; Birkland, T.; Chen, P.; Duffield, J.; Schnapp, L.M.; et al. CYR61 (CCN1) overexpression induces lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L759–L765. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Lau, L.F. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009, 41, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Son, S.; Shin, I. Role of the CCN protein family in cancer. BMB Rep. 2018, 51, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Henrot, P.; Truchetet, M.E.; Fisher, G.; Taïeb, A.; Cario, M. CCN proteins as potential actionable targets in scleroderma. Exp. Dermatol. 2019, 28, 11–18. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Kurundkar, A.R.; Kurundkar, D.; Rangarajan, S.; Locy, M.L.; Zhou, Y.; Liu, R.M.; Zmijewski, J.; Thannickal, V.J. The matricellular protein CCN1 enhances TGF-β1/SMAD3-dependent profibrotic signaling in fibroblasts and contributes to fibrogenic responses to lung injury. Faseb J. 2016, 30, 2135–2150. [Google Scholar] [CrossRef]
- Kulkarni, T.; Kurundkar, A.R.; Kim, Y.I.; de Andrade, J.; Luckhardt, T.; Thannickal, V.J. The senescence-associated matricellular protein CCN1 in plasma of human subjects with idiopathic pulmonary fibrosis. Respir. Med. 2020, 161, 105821. [Google Scholar] [CrossRef]
- Hu, L.; Yu, Y.; Huang, H.; Fan, H.; Hu, L.; Yin, C.; Li, K.; Fulton, D.J.; Chen, F. Epigenetic Regulation of Interleukin 6 by Histone Acetylation in Macrophages and Its Role in Paraquat-Induced Pulmonary Fibrosis. Front. Immunol. 2016, 7, 696. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Pedroza, M.; Schneider, D.J.; Karmouty-Quintana, H.; Coote, J.; Shaw, S.; Corrigan, R.; Molina, J.G.; Alcorn, J.L.; Galas, D.; Gelinas, R.; et al. Interleukin-6 Contributes to Inflammation and Remodeling in a Model of Adenosine Mediated Lung Injury. PLoS ONE 2011, 6, e22667. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.S.; Mishra, V.; Shukla, M.; Verma, M.; Chaudhury, B.P.; Kumar, P.; Chhabra, J.K.; Pandey, H.P.; Paul, B. IL-6 receptor-mediated lung Th2 cytokine networking in silica-induced pulmonary fibrosis. Arch. Toxicol. 2010, 84, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.-M.; Tseng, H.-Y.; Jung, F.; Yang, S.-H.; Lin, J.-C. Elevation of IL-6 and IL-33 Levels in Serum Associated with Lung Fibrosis and Skeletal Muscle Wasting in a Bleomycin-Induced Lung Injury Mouse Model. Mediat. Inflamm. 2019, 2019, 7947596. [Google Scholar] [CrossRef]
- Saito, F.; Tasaka, S.; Inoue, K.; Miyamoto, K.; Nakano, Y.; Ogawa, Y.; Yamada, W.; Shiraishi, Y.; Hasegawa, N.; Fujishima, S.; et al. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am. J. Respir. Cell Mol. Biol. 2008, 38, 566–571. [Google Scholar] [CrossRef]
- Le, T.T.; Karmouty-Quintana, H.; Melicoff, E.; Le, T.T.; Weng, T.; Chen, N.Y.; Pedroza, M.; Zhou, Y.; Davies, J.; Philip, K.; et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J. Immunol. 2014, 193, 3755–3768. [Google Scholar] [CrossRef]
- Kida, H.; Yoshida, M.; Hoshino, S.; Inoue, K.; Yano, Y.; Yanagita, M.; Kumagai, T.; Osaki, T.; Tachibana, I.; Saeki, Y.; et al. Protective effect of IL-6 on alveolar epithelial cell death induced by hydrogen peroxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L342–L349. [Google Scholar] [CrossRef] [PubMed]
- Maleki, F.; Ovens, K.; McQuillan, I.; Kusalik, A.J. Size matters: How sample size affects the reproducibility and specificity of gene set analysis. Hum. Genom. 2019, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, A.; Tosco-Herrera, E.; Molina-Molina, M.; Flores, C. Idiopathic pulmonary fibrosis and the role of genetics in the era of precision medicine. Front. Med. 2023, 10, 1152211. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Sadroddiny, E. MicroRNAs in lung diseases: Recent findings and their pathophysiological implications. Pulm. Pharmacol. Ther. 2015, 34, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ding, Y.; Hou, Y.; Yu, T.; Nie, H.; Cui, Y. The miR-130a-3p/TGF-βRII Axis Participates in Inhibiting the Differentiation of Fibroblasts Induced by TGF-β1. Front. Pharmacol. 2021, 12, 732540. [Google Scholar] [CrossRef]
- Lino Cardenas, C.L.; Henaoui, I.S.; Courcot, E.; Roderburg, C.; Cauffiez, C.; Aubert, S.; Copin, M.C.; Wallaert, B.; Glowacki, F.; Dewaeles, E.; et al. miR-199a-5p Is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS Genet. 2013, 9, e1003291. [Google Scholar] [CrossRef]
- Das, S.; Kumar, M.; Negi, V.; Pattnaik, B.; Prakash, Y.S.; Agrawal, A.; Ghosh, B. MicroRNA-326 Regulates Profibrotic Functions of Transforming Growth Factor-β in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 882–892. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, G.; Kang, Y.; Dong, Z.; Qian, Q.; Ma, X. N-Cadherin Expression Is Associated with Acquisition of EMT Phenotype and with Enhanced Invasion in Erlotinib-Resistant Lung Cancer Cell Lines. PLoS ONE 2013, 8, e57692. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Yoshino, I.; Yamaguchi, R.; Shimamura, T.; Nagasaki, M.; Imoto, S.; Niida, A.; Koizumi, F.; Kohno, T.; Yokota, J.; et al. N-cadherin expression is a potential survival mechanism of gefitinib-resistant lung cancer cells. Am. J. Cancer Res. 2011, 1, 823–833. [Google Scholar]
- Tang, H.; Chen, J.; Han, X.; Feng, Y.; Wang, F. Upregulation of SPP1 Is a Marker for Poor Lung Cancer Prognosis and Contributes to Cancer Progression and Cisplatin Resistance. Front. Cell Dev. Biol. 2021, 9, 646390. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Naba, A.; Bhutkar, A.; Guardia, T.; Miller, K.M.; Li, C.M.; Dayton, T.L.; Sanchez-Rivera, F.J.; Kim-Kiselak, C.; Jailkhani, N.; et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. USA 2017, 114, E5625–E5634. [Google Scholar] [CrossRef]
- Boccellino, M.; Pinto, F.; Ieluzzi, V.; Giovane, A.; Quagliuolo, L.; Fariello, C.; Coppola, M.; Carlucci, A.; Santini, M.; Ferati, K.; et al. Proteomics analysis of human serum of patients with non-small-cell lung cancer reveals proteins as diagnostic biomarker candidates. J. Cell Physiol. 2019, 234, 23798–23806. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Sun, F.; Zhou, J.; Li, L.; Shapiro, S.D.; Xiao, G. Interleukin-6 Prevents the Initiation but Enhances the Progression of Lung Cancer. Cancer Res. 2015, 75, 3209–3215. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | Platform | Samples | Reference | |
|---|---|---|---|---|
| Control | IPF | |||
| GSE24206 | GPL570 | 6 | 17 | [19] |
| GSE21369 | GPL570 | 6 | 11 | [20] |
| GSE110147 | GLP6244 | 11 | 22 | [21] |
| GSE72073 | GPL6480 | 3 | 5 | [22] |
| GSE32539 | GPL6244 | 50 | 119 | [23] |
| Total | 76 | 174 | ||
| Category | Term | Count | p-Value | Genes |
|---|---|---|---|---|
| BP | GO:0030198~extracellular matrix organization | 5 | 2.17 × 10−6 | SPP1, TNC, FN1, CYR61, FBN1 |
| BP | GO:0007155~cell adhesion | 5 | 6.23 × 10−5 | CDH2, SPP1, TNC, FN1, CYR61 |
| BP | GO:0006953~acute-phase response | 3 | 1.87 × 10−4 | IL6, SERPINA1, FN1 |
| BP | GO:0022617~extracellular matrix disassembly | 3 | 7.13 × 10−4 | SPP1, FN1, FBN1 |
| BP | GO:0042060~wound healing | 3 | 7.90 × 10−4 | IL6, TNC, FN1 |
| MF | GO:0008201~heparin binding | 4 | 6.73 × 10−5 | FN1, APOE, CYR61, FBN1 |
| MF | GO:0005178~integrin binding | 3 | 0.00134085 | FN1, CYR61, FBN1 |
| MF | GO:0050840~extracellular matrix binding | 2 | 0.01377989 | SPP1, CYR61 |
| CC | GO:0005576~extracellular region | 9 | 3.03 × 10−8 | IL6, TF, SERPINA1, SPP1, TNC, FN1, APOE, CYR61, FBN1 |
| CC | GO:0005615~extracellular space | 8 | 3.74 × 10−7 | IL6, TF, SERPINA1, SPP1, TNC, FN1, APOE, FBN1 |
| CC | GO:0031012~extracellular matrix | 5 | 8.06 × 10−6 | TNC, FN1, APOE, CYR61, FBN1 |
| CC | GO:0005578~proteinaceous extracellular matrix | 4 | 2.47 × 10−4 | SERPINA1, FN1, CYR61, FBN1 |
| CC | GO:0070062~extracellular exosome | 7 | 7.38 × 10−4 | TF, SERPINA1, CDH2, SPP1, FN1, APOE, FBN1 |
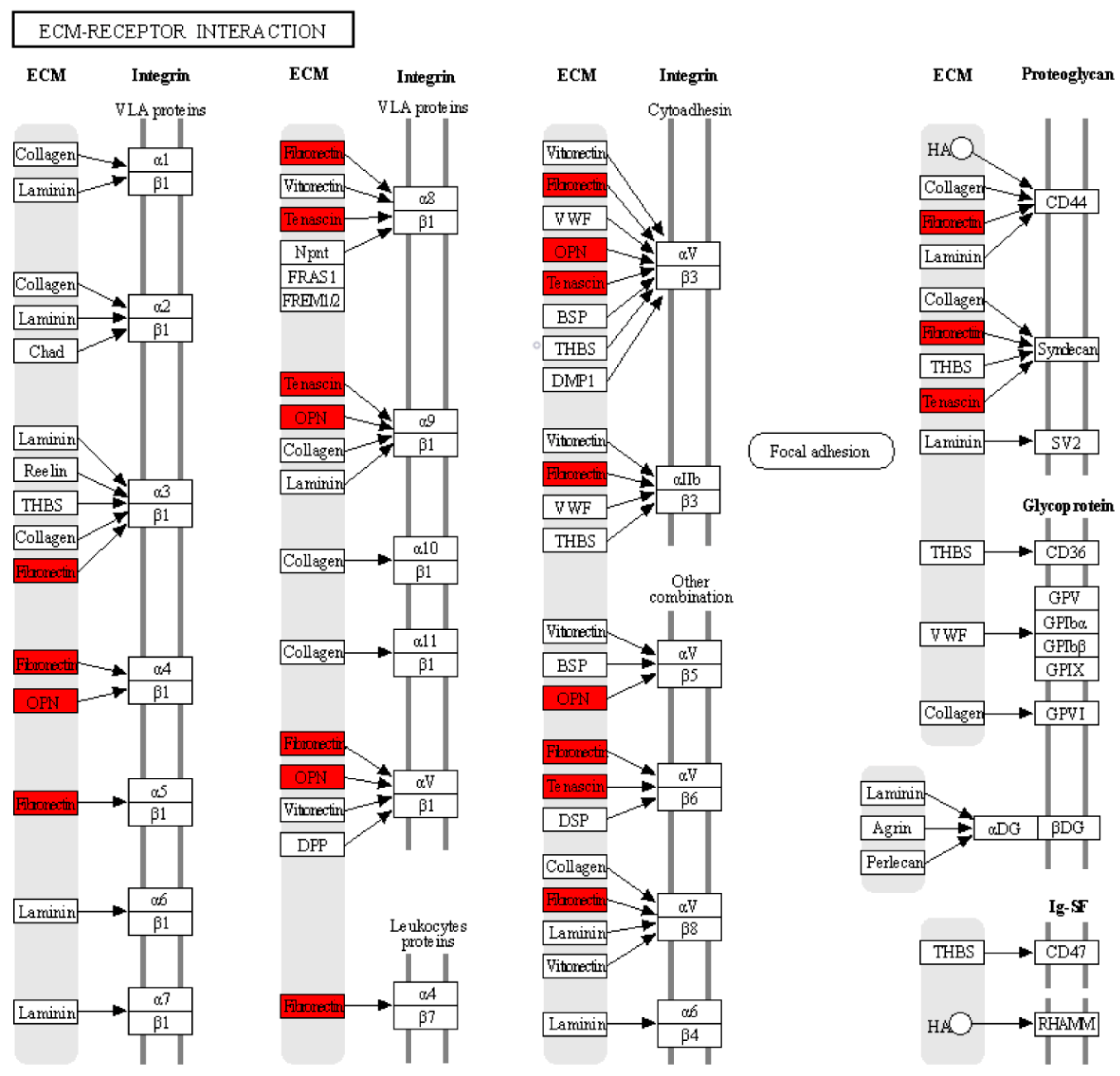
| KEGG | hsa04512: ECM–receptor interaction | 3 | 0.00318652 | SPP1, TNC, FN1 |
| KEGG | hsa04151: PI3K-Akt signaling pathway | 4 | 0.00376323 | IL6, SPP1, TNC, FN1 |
| KEGG | hsa04510: Focal adhesion | 3 | 0.01697039 | SPP1, TNC, FN1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velázquez-Enríquez, J.M.; Reyes-Avendaño, I.; Santos-Álvarez, J.C.; Reyes-Jiménez, E.; Vásquez-Garzón, V.R.; Baltiérrez-Hoyos, R. Identification of Hub Genes in Idiopathic Pulmonary Fibrosis and Their Association with Lung Cancer by Bioinformatics Analysis. Adv. Respir. Med. 2023, 91, 407-431. https://doi.org/10.3390/arm91050032
Velázquez-Enríquez JM, Reyes-Avendaño I, Santos-Álvarez JC, Reyes-Jiménez E, Vásquez-Garzón VR, Baltiérrez-Hoyos R. Identification of Hub Genes in Idiopathic Pulmonary Fibrosis and Their Association with Lung Cancer by Bioinformatics Analysis. Advances in Respiratory Medicine. 2023; 91(5):407-431. https://doi.org/10.3390/arm91050032
Chicago/Turabian StyleVelázquez-Enríquez, Juan Manuel, Itayetzi Reyes-Avendaño, Jovito Cesar Santos-Álvarez, Edilburga Reyes-Jiménez, Verónica Rocío Vásquez-Garzón, and Rafael Baltiérrez-Hoyos. 2023. "Identification of Hub Genes in Idiopathic Pulmonary Fibrosis and Their Association with Lung Cancer by Bioinformatics Analysis" Advances in Respiratory Medicine 91, no. 5: 407-431. https://doi.org/10.3390/arm91050032
APA StyleVelázquez-Enríquez, J. M., Reyes-Avendaño, I., Santos-Álvarez, J. C., Reyes-Jiménez, E., Vásquez-Garzón, V. R., & Baltiérrez-Hoyos, R. (2023). Identification of Hub Genes in Idiopathic Pulmonary Fibrosis and Their Association with Lung Cancer by Bioinformatics Analysis. Advances in Respiratory Medicine, 91(5), 407-431. https://doi.org/10.3390/arm91050032