Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations?



Abstract
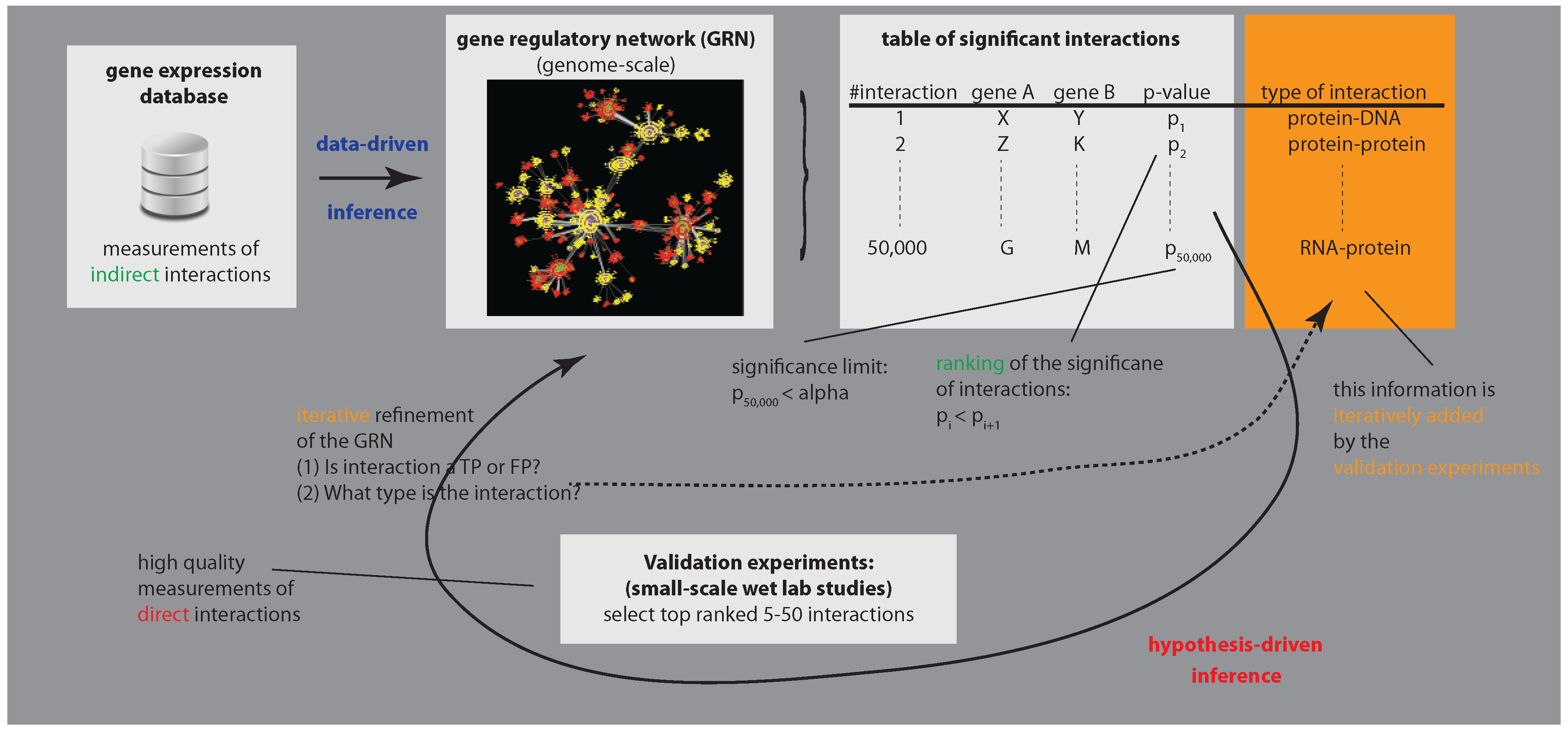
1. Introduction
2. How Large Are Gene Regulatory Networks?
3. Biological Validation of GRNs
3.1. Enhancing Experimental Assays by Perturbing the System
- A single gene knockdown experiment was selected from the collection that included all replicates as a validation set.
- The genes whose expressions are significantly affected by the perturbation were identified.
- The capacity of the network model to predict which genes are affected by the perturbation by focusing on connections local to the gene being perturbed was assessed.
- Steps 1–3 were repeated to assess the predictive power of the network model until all perturbations had been tested.
3.2. Existing Data Repositories
4. General Considerations about Validation
- A validation is necessary because the entities to be validated are generated by a statistical model corresponding to its predictions.
- The entities to be predicted are scientifically meaningful for particular research fields.
5. Clinical Validation of GRNs
Higher-Level Networks
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ghahramani, Z. Probabilistic machine learning and artificial intelligence. Nature 2015, 521, 452. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436. [Google Scholar] [CrossRef] [PubMed]
- Witten, D.M.; Friedman, J.H.; Simon, N. New Insights and Faster Computations for the Graphical Lasso. J. Comput. Graph. Stat. 2011, 20, 892–900. [Google Scholar] [CrossRef]
- De Matos Simoes, R.; Emmert-Streib, F. Bagging statistical network inference from large-scale gene expression data. PLoS ONE 2012, 7, e33624. [Google Scholar] [CrossRef] [PubMed]
- Opgen-Rhein, R.; Strimmer, K. Learning causal networks from systems biology time course data: An effective model selection procedure for the vector autoregressive process. BMC Bioinform. 2007, 8, S3. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-Generation DNA Sequencing Methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, J.; Wyrwicz, L.S. Integrating genomics, proteomics and bioinformatics in translational studies of molecular medicine. Expert Rev. Mol. Diagn. 2009, 9, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Oltvai, Z.N. Network Biology: Understanding the Cell’s Functional Organization. Nat. Rev. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Lee, E.; Liu, Y.T.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Dehmer, M.; Jurisica, I. Knowledge discovery and interactive data mining in bioinformatics-state-of-the-art, future challenges and research directions. BMC Bioinform. 2014, 15, I1. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Hidalgo, D.; Domínguez-Cejudo, M.A.; Amore, G.; Brockmann, A.; Lemos, M.C.; Córdoba, A.; Casares, F. A Hh-driven gene network controls specification, pattern and size of the Drosophila simple eyes. Development 2012, 082172. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Hidalgo, D.; Zurita, A.C.; Fernández, M.C.L. Complex networks evolutionary dynamics using genetic algorithms. Int. J. Bifurc. Chaos 2012, 22, 1250156. [Google Scholar] [CrossRef]
- Aguilar-Hidalgo, D.; Lemos, M.C.; Córdoba, A. Evolutionary dynamics in gene networks and inference algorithms. Computation 2015, 3, 99–113. [Google Scholar] [CrossRef]
- Ideker, T.; Krogan, N.J. Differential network biology. Mol. Syst. Biol. 2012, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulatory network of Escherichia coli. Nat. Genet. 2002, 31, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Roukos, D.H. Genome network medicine: Innovation to overcome huge challenges in cancer therapy. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Yachie-Kinoshita, A.; Onishi, K.; Ostblom, J.; Langley, M.A.; Posfai, E.; Rossant, J.; Zandstra, P.W. Modeling signaling-dependent pluripotency with Boolean logic to predict cell fate transitions. Mol. Syst. Biol. 2018, 14, e7952. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, E.R. Validation of inference procedures for gene regulatory networks. Curr. Genom. 2007, 8, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, E.R. Validation of gene regulatory networks: Scientific and inferential. Brief. Bioinform. 2011, 12, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A. What does biologically meaningful mean? A perspective on gene regulatory network validation. Genome Biol. 2011, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Schaffter, T.; Marbach, D.; Floreano, D. GeneNetWeaver: In silico benchmark generation and performance profiling of network inference methods. Bioinformatics 2011, 27, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Van den Bulcke, T.; Van Leemput, K.; Naudts, B.; van Remortel, P.; Ma, H.; Verschoren, A.; De Moor, B.; Marchal, K. SynTReN: A generator of synthetic gene expression data for design and analysis of structure learning algorithms. BMC Bioinform. 2006, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; Glazko, G.; Altay, G.; de Matos Simoes, R. Statistical inference and reverse engineering of gene regulatory networks from observational expression data. Front. Genet. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Pearl, J. Causality: Models, Reasoning, and Inference; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2000. [Google Scholar]
- Spirtes, P.; Glymour, C.; Scheines, R. Causation, Prediction, and Search; Springer: New York, NY, USA, 1993. [Google Scholar]
- Emmert-Streib, F. The Chronic Fatigue Syndrome: A Comparative Pathway Analysis. J. Comput. Biol. 2007, 14, 961–972. [Google Scholar] [CrossRef] [PubMed]
- De Matos Simoes, R.; Dehmer, M.; Emmert-Streib, F. Interfacing cellular networks of S. cerevisiae and E. coli: Connecting dynamic and genetic information. BMC Genom. 2013, 14, 324. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse Engineering of Regulatory Networks in Human B Cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef] [PubMed]
- De Matos Simoes, R.; Dehmer, M.; Emmert-Streib, F. B-cell lymphoma gene regulatory networks: Biological consistency among inference methods. Front. Genet. 2013, 4, 281. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; de Matos Simoes, R.; Mullan, P.; Haibe-Kains, B.; Dehmer, M. The gene regulatory network for breast cancer: Integrated regulatory landscape of cancer hallmarks. Front. Genet. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; de Matos Simoes, R.; Glazko, G.; McDade, S.; Haibe-Kains, B.; Holzinger, A.; Dehmer, M.; Campbell, F. Functional and genetic analysis of the colon cancer network. BMC Bioinform. 2014, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Godfrey-Smith, P. Theory and Reality: An Introduction to the Philosophy of Science; Science and Its Conceptual Foundations Series; University of Chicago Press: Chicago, IL, USA, 2003. [Google Scholar]
- Kell, D.B.; Oliver, S.G. Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. BioEssays 2004, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Bruce, J. Chemical Cross-Linking for Protein? Protein Interaction Studies. In Mass Spectrometry of Proteins and Peptides; Methods in Molecular Biology; Lipton, M., Pasa-Tolic, L., Eds.; Humana Press: New York, NY, USA, 2009; Volume 492, pp. 283–293. [Google Scholar]
- Liu, Q.; Dinu, I.; Adewale, A.; Potter, J.; Yasui, Y. Comparative evaluation of gene-set analysis methods. BMC Bioinform. 2007, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.G.; Roth, S.Y. Current Protocols in Molecular Biology; Chapter Identification of Protein Interactions by Far Western Analysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2009, 38, D525–D531. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.; Lieb, J. ChIP-chip: Considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 2004, 83, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kidder, B.L.; Hu, G.; Zhao, K. ChIP-Seq: Technical considerations for obtaining high-quality data. Nat. Immunol. 2011, 12, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Seeholzer, S.; Ohh, M. Identification of associated proteins by coimmunoprecipitation. In Molecular Cloning—A Laboratory Manual; Sambrook, J., Russell, D.W., Eds.; CSHL Press: New York, NY, USA, 2002; Volume 3, pp. 18–60. [Google Scholar]
- Joung, J.K.; Ramm, E.I.; Pabo, C.O. A bacterial two-hybrid selection system for studying protein–DNA and protein–protein interactions. Proc. Natl. Acad. Sci. USA 2000, 97, 7382–7387. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J. Vis. Exp. 2011, 50, 2638. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.; Valle, D.; McKusick, V.A. Online Mendelian inheritance in man (OMIM). Hum. Mutat. 2000, 15, 57–61. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.; He, X.; Perou, C. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Haibe-Kains, B.; Desmedt, C.; Loi, S.; Culhane, A.C.; Bontempi, G.; Quackenbush, J.; Sotiriou, C. A Three-Gene Model to Robustly Identify Breast Cancer Molecular Subtypes. J. Natl. Cancer Inst. 2012, 104, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Olsen, C.; Fleming, K.; Prendergast, N.; Rubio, R.; Emmert-Streib, F.; Bontempi, G.; Haibe-Kains, B.; Quackenbush, J. Inference and validation of predictive gene networks from biomedical literature and gene expression data. Genomics 2014, 103, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2017, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hamo, R.; Efroni, S. Gene expression and network-based analysis reveals a novel role for hsa-miR-9 and drug control over the p38 network in glioblastoma multiforme progression. Genome Med. 2011, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, M.; Mueller, L.; Emmert-Streib, F. Quantitative Network Measures as Biomarkers for Classifying Prostate Cancer Disease States: A Systems Approach to Diagnostic Biomarkers. PLoS ONE 2013, 8, e77602. [Google Scholar] [CrossRef] [PubMed]
- Cun, Y.; Frohlich, H. Network and Data Integration for Biomarker Signature Discovery via Network Smoothed T-Statistics. PLoS ONE 2013, 8, e73074. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, H. Including network knowledge into Cox regression models for biomarker signature discovery. Biom. J. 2014, 56, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, G.; Chen, L. Prediction and early diagnosis of complex diseases by edge-network. Bioinformatics 2014, 30, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, W.; Yu, X.; Liu, X.; Li, M.; Liu, R.; Chen, L. Edge biomarkers for classification and prediction of phenotypes. Sci. China Life Sci. 2014, 57, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Sun, S.y.; Wang, Y.; Zhu, H.; Chen, L. Network biomarkers reveal dysfunctional gene regulations during disease progression. FEBS J. 2013, 280, 5682–5695. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Q.; Tingle, S.R.; Filipski, K.K.; Khoury, M.J.; Lam, T.K.; Schully, S.D.; Ioannidis, J.P. An overview of recommendations and translational milestones for genomic tests in cancer. Genet. Med. 2014, 17, 431. [Google Scholar] [CrossRef] [PubMed]
- Van De Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Castiglioni, I. In silico identification of drug target pathways in breast cancer subtypes using pathway cross-talk inhibition. J. Transl. Med. 2018, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, S.; Igea, A.; Arroyo, R.; Alcalde, V.; Canovas, B.; Orozco, M.; Nebreda, A.R.; Aloy, P. Quantification of pathway cross-talk reveals novel synergistic drug combinations for breast cancer. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Organism (Network Type) | # of Interactions | References |
|---|---|---|
| E. coli (GRN) | 21,820 | [28] |
| S. cerevisiae (GRN) | 27,493 | [4] |
| B-cell lymphoma (GRN) | 129,000; 57,905 | [29,30] |
| Breast cancer (GRN) | 180,171 | [31] |
| Colon cancer (GRN) | 135,194 | [32] |
| S. cerevisiae (TRN) | 12,873 | [28] |
| S. cerevisiae (PIN) | 112,562 | [28] |
| Human (TRN) | 51,871 | [30] |
| Human (PIN) | 185,433 | [30] |
| Experimental Technique | Type of Interaction | Reference |
|---|---|---|
| ChIP-chip/ ChIP-seq | protein–DNA interaction | [40,41] |
| Co-immunoprecipitation | protein–protein interaction | [42] |
| Yeast two-hybrid | protein–protein interaction | [43] |
| Crosslinking Protein Interaction Analysis | protein–protein interaction | [35] |
| Label Transfer Chemistry | protein–protein interaction | [36] |
| Far-Western Blot Analysis | protein–protein interaction | [37] |
| BiFCassays | protein–protein interaction | [38] |
| iCLIP | protein–RNA interaction | [44] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emmert-Streib, F.; Dehmer, M. Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations? Mach. Learn. Knowl. Extr. 2019, 1, 138-148. https://doi.org/10.3390/make1010008
Emmert-Streib F, Dehmer M. Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations? Machine Learning and Knowledge Extraction. 2019; 1(1):138-148. https://doi.org/10.3390/make1010008
Chicago/Turabian StyleEmmert-Streib, Frank, and Matthias Dehmer. 2019. "Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations?" Machine Learning and Knowledge Extraction 1, no. 1: 138-148. https://doi.org/10.3390/make1010008
APA StyleEmmert-Streib, F., & Dehmer, M. (2019). Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations? Machine Learning and Knowledge Extraction, 1(1), 138-148. https://doi.org/10.3390/make1010008