Compensation of Water Influence on Anionic Polymerization of ε-Caprolactam: 1. Chemistry and Experiments


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods for Manufacturing
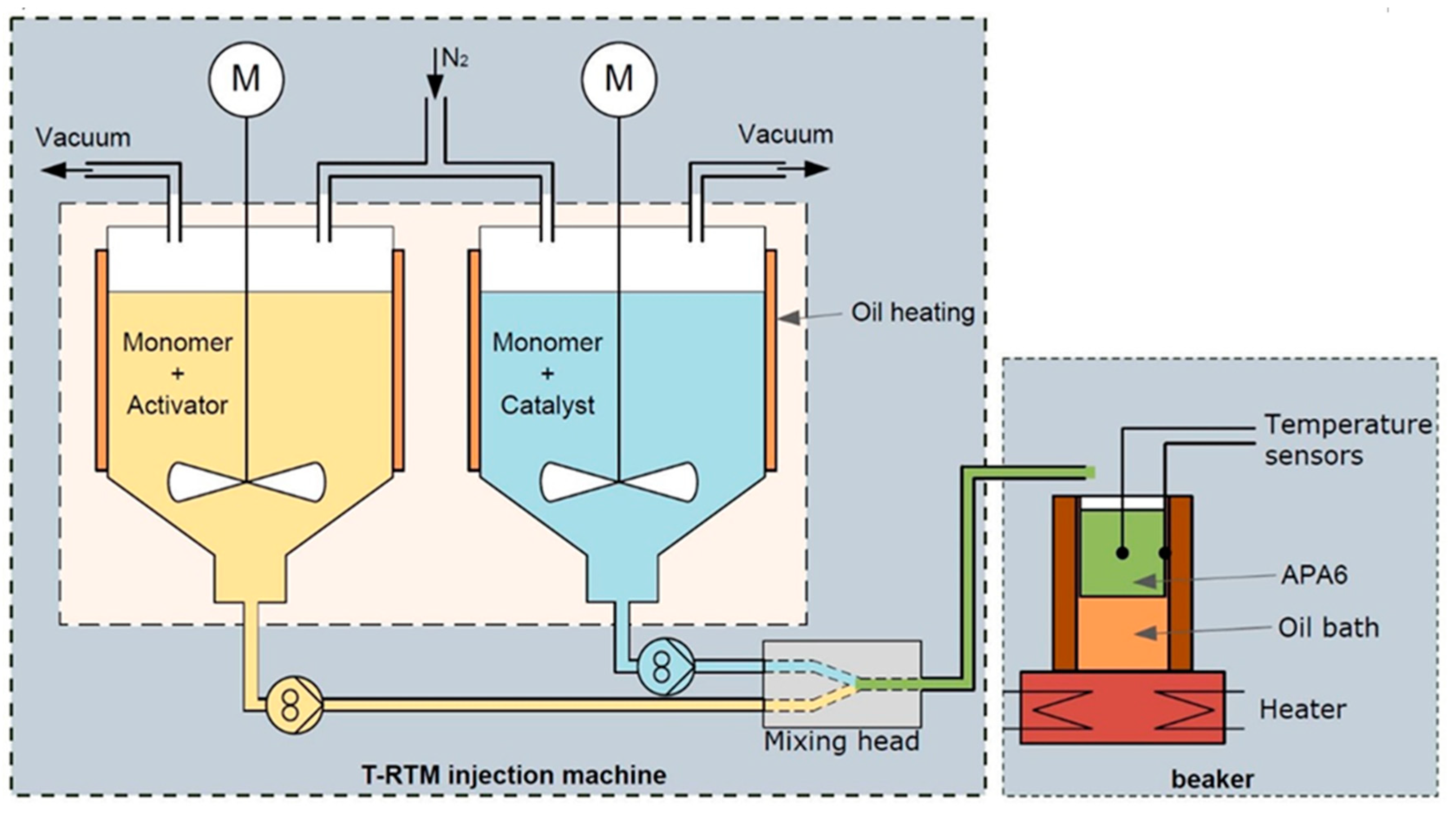
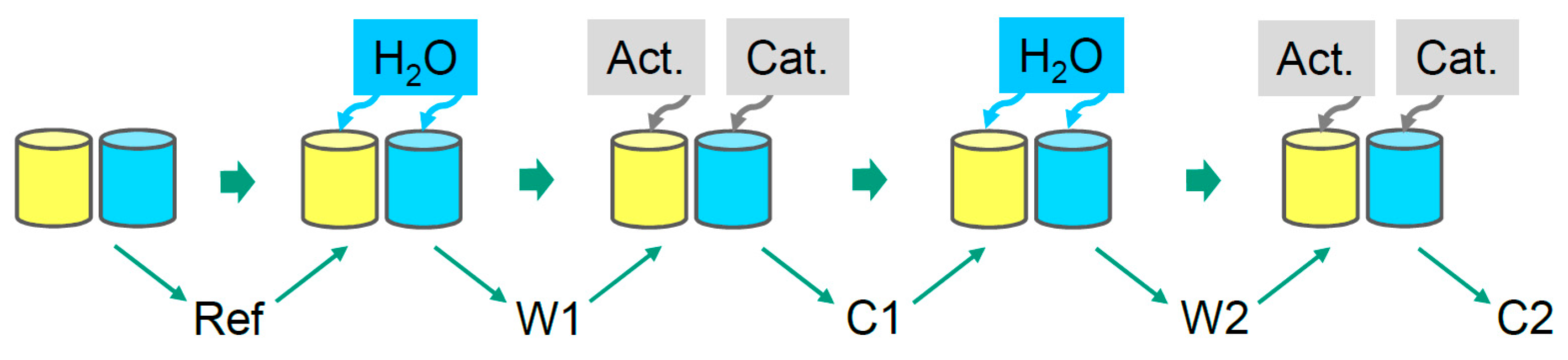
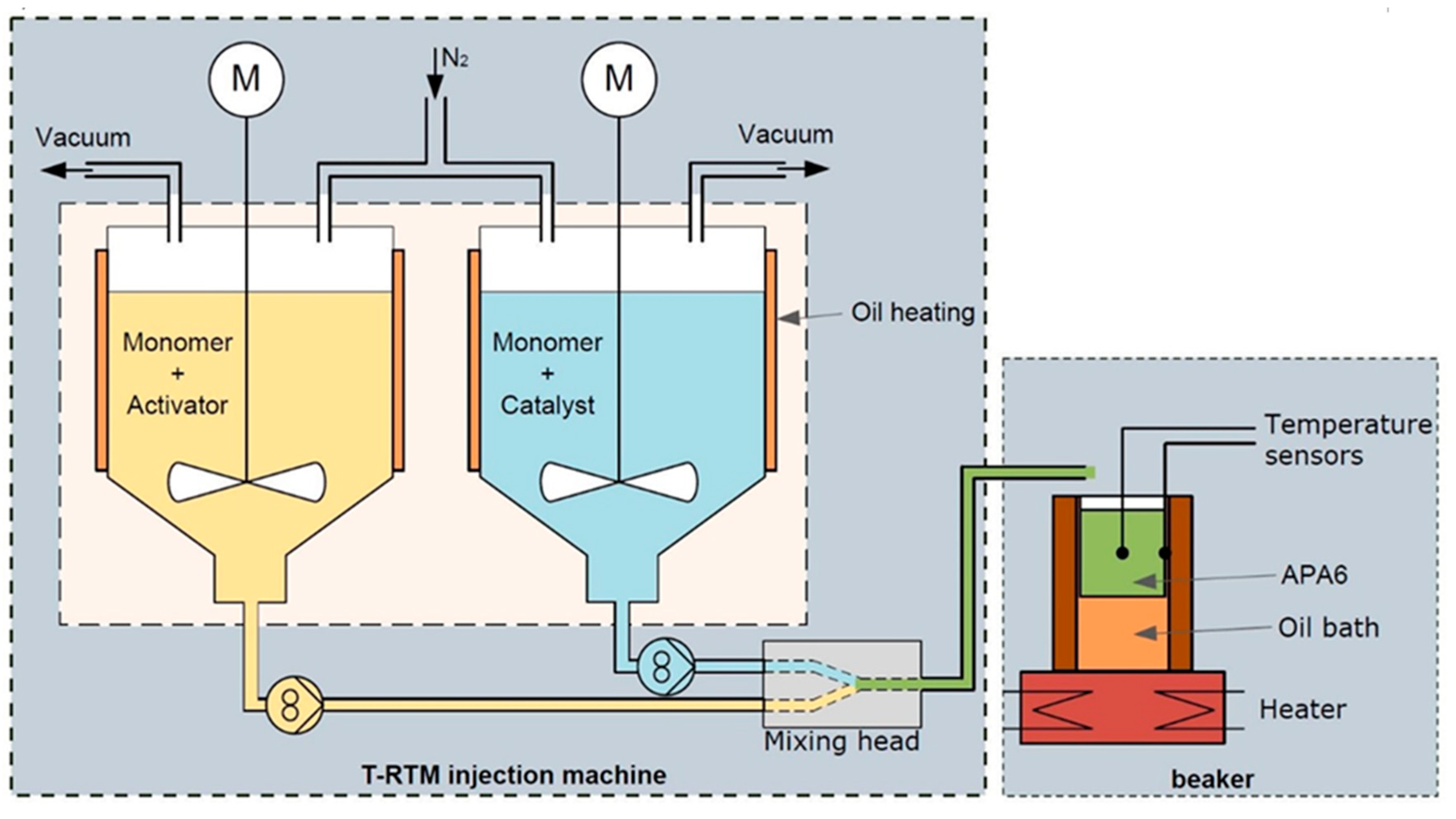
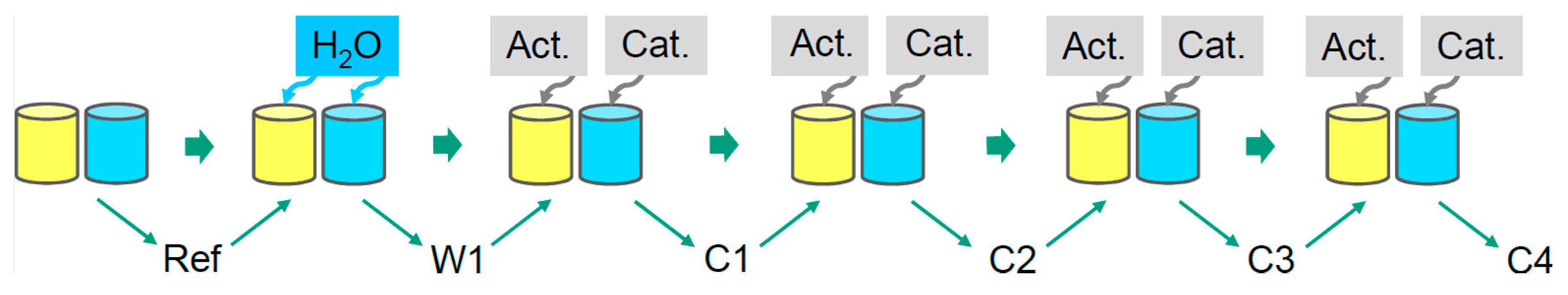
2.2.1. Cure Monitoring with Beaker
2.2.2. Plate Manufacturing
2.3. Methods for Characterization
2.3.1. Mechanical Properties
2.3.2. Differential Scanning Calorimetry
2.3.3. Residual Monomer Content
2.3.4. Viscosity Average Molecular Mass
2.3.5. Average Molecular Mass
3. Results and Discussion
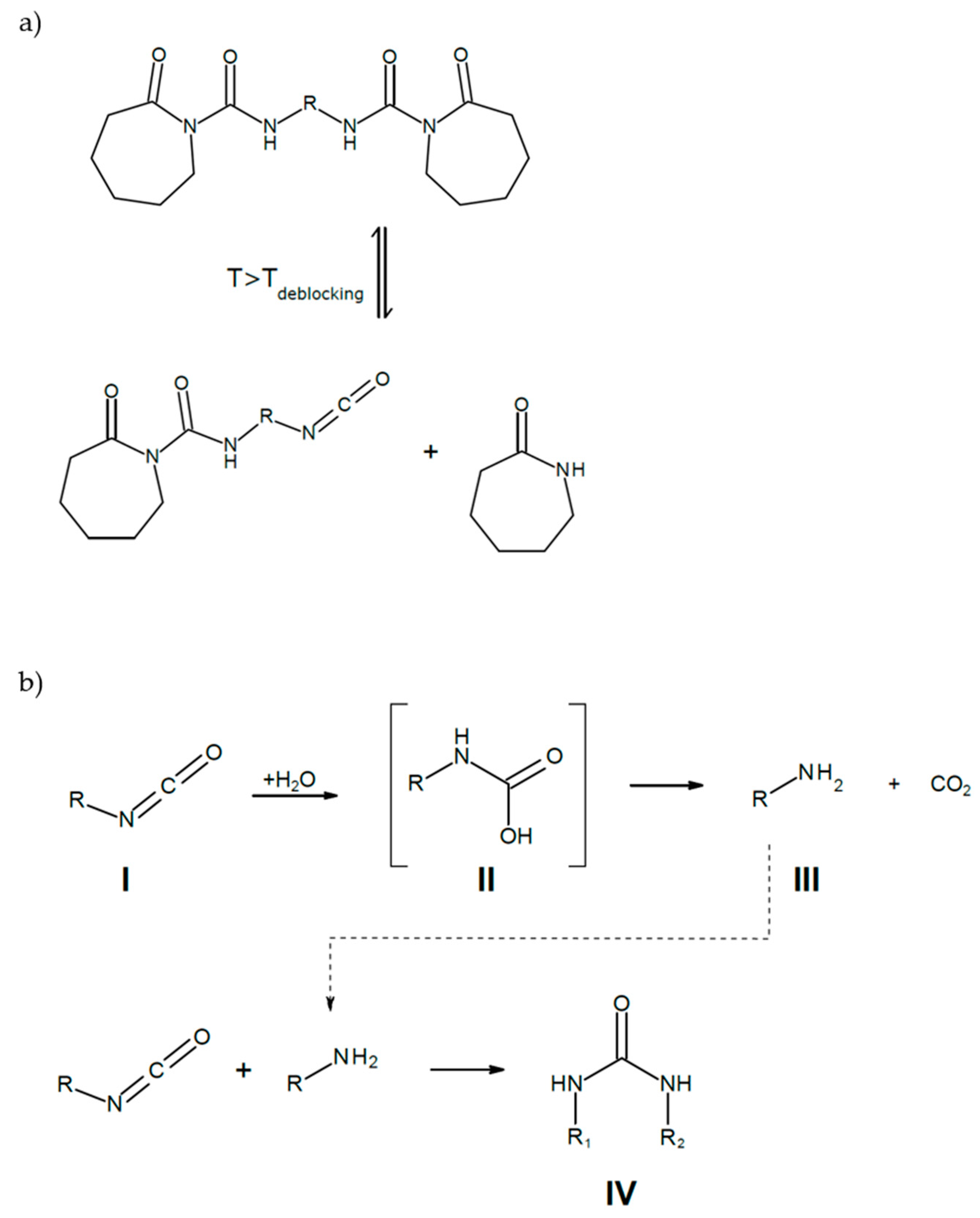
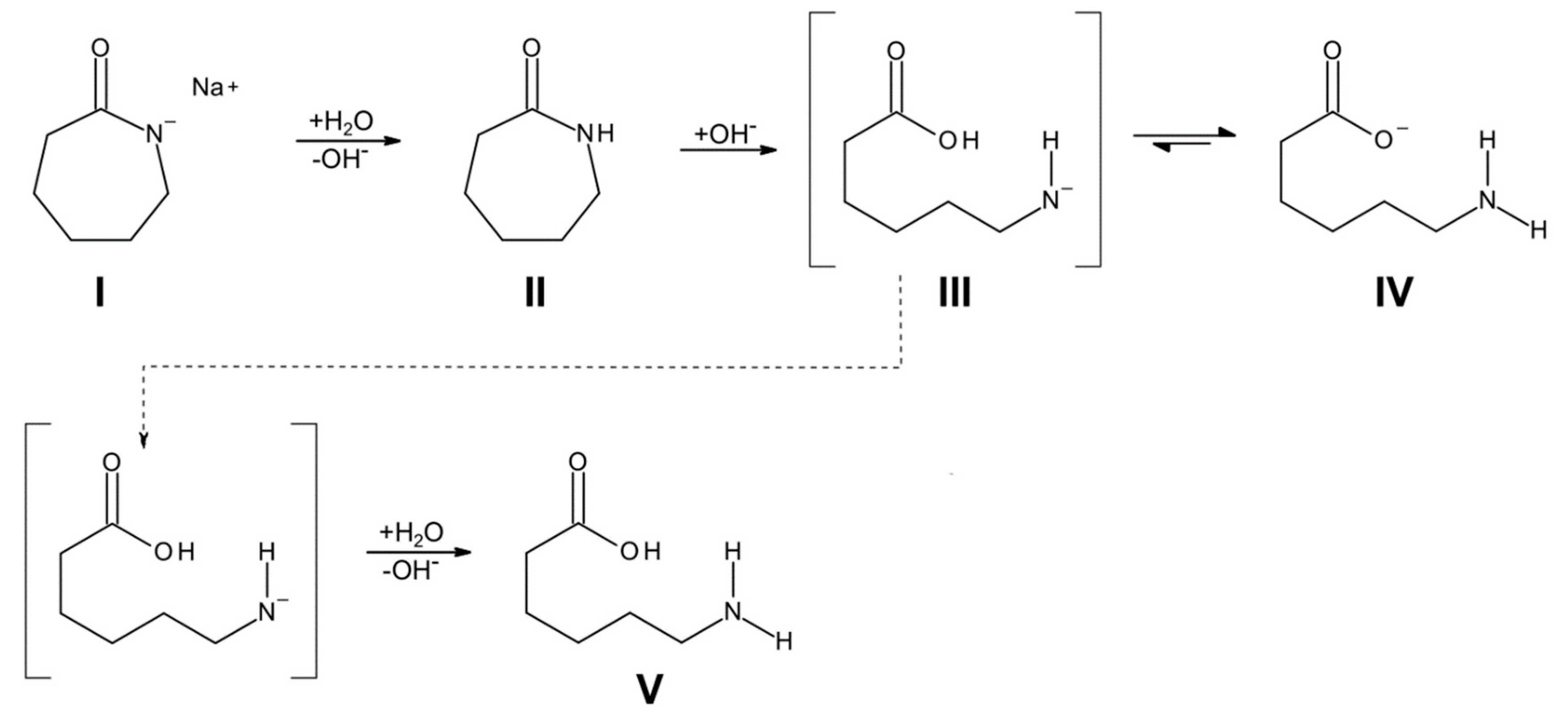
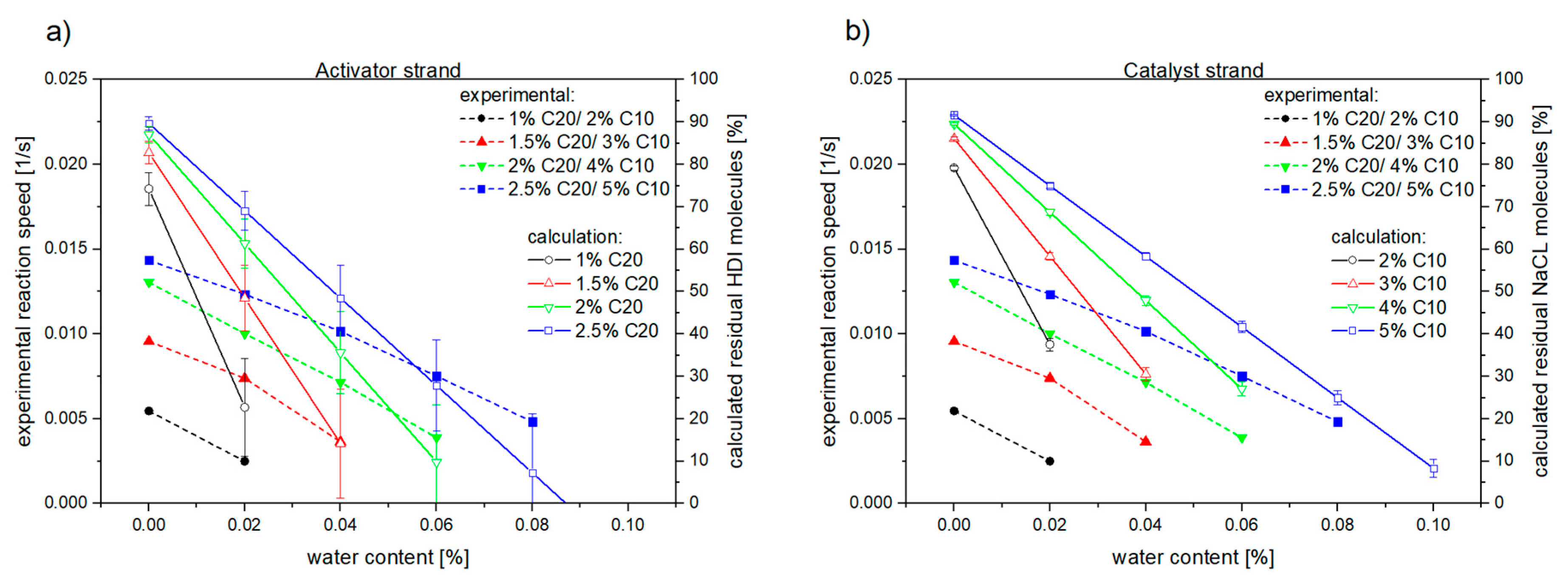
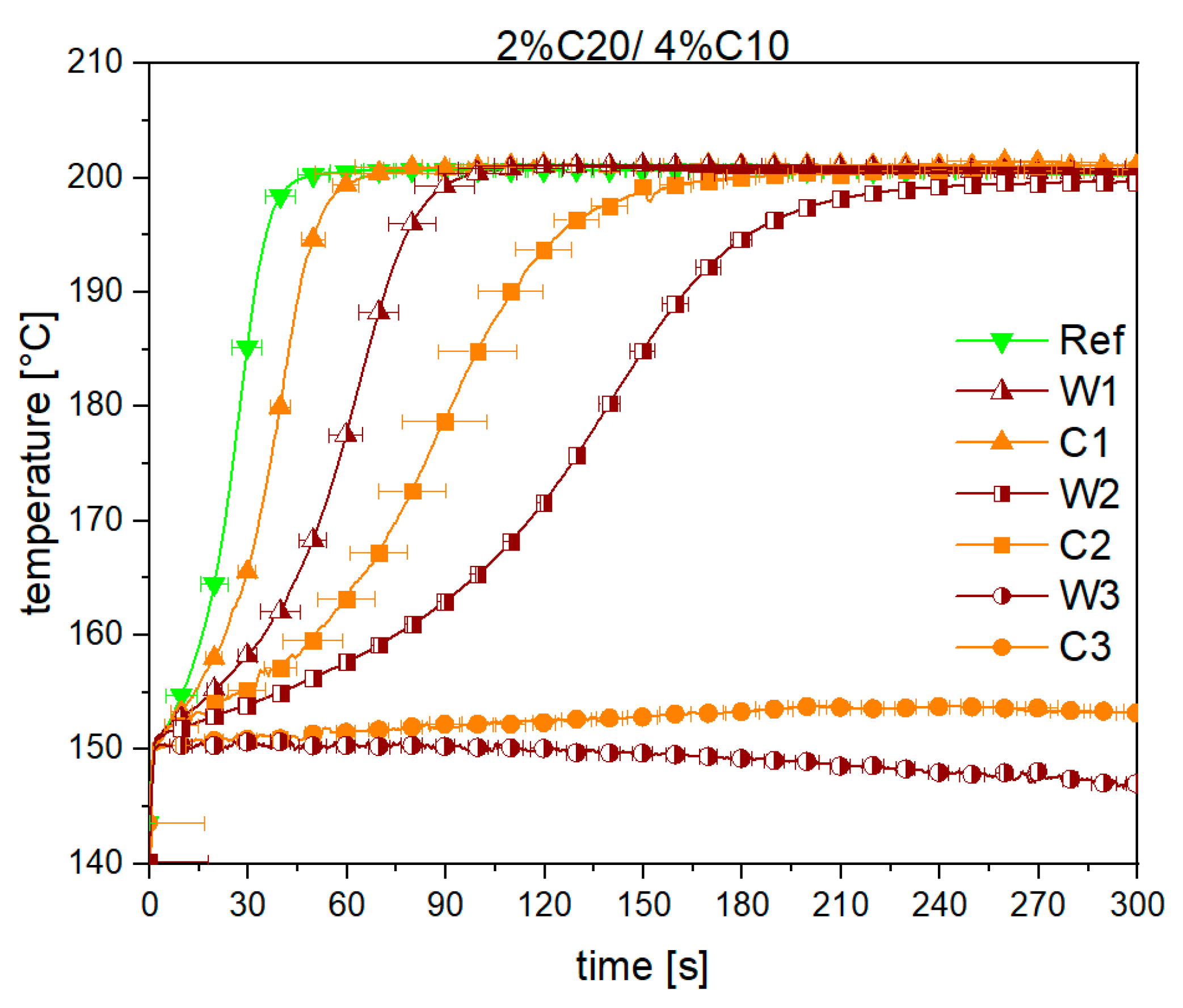
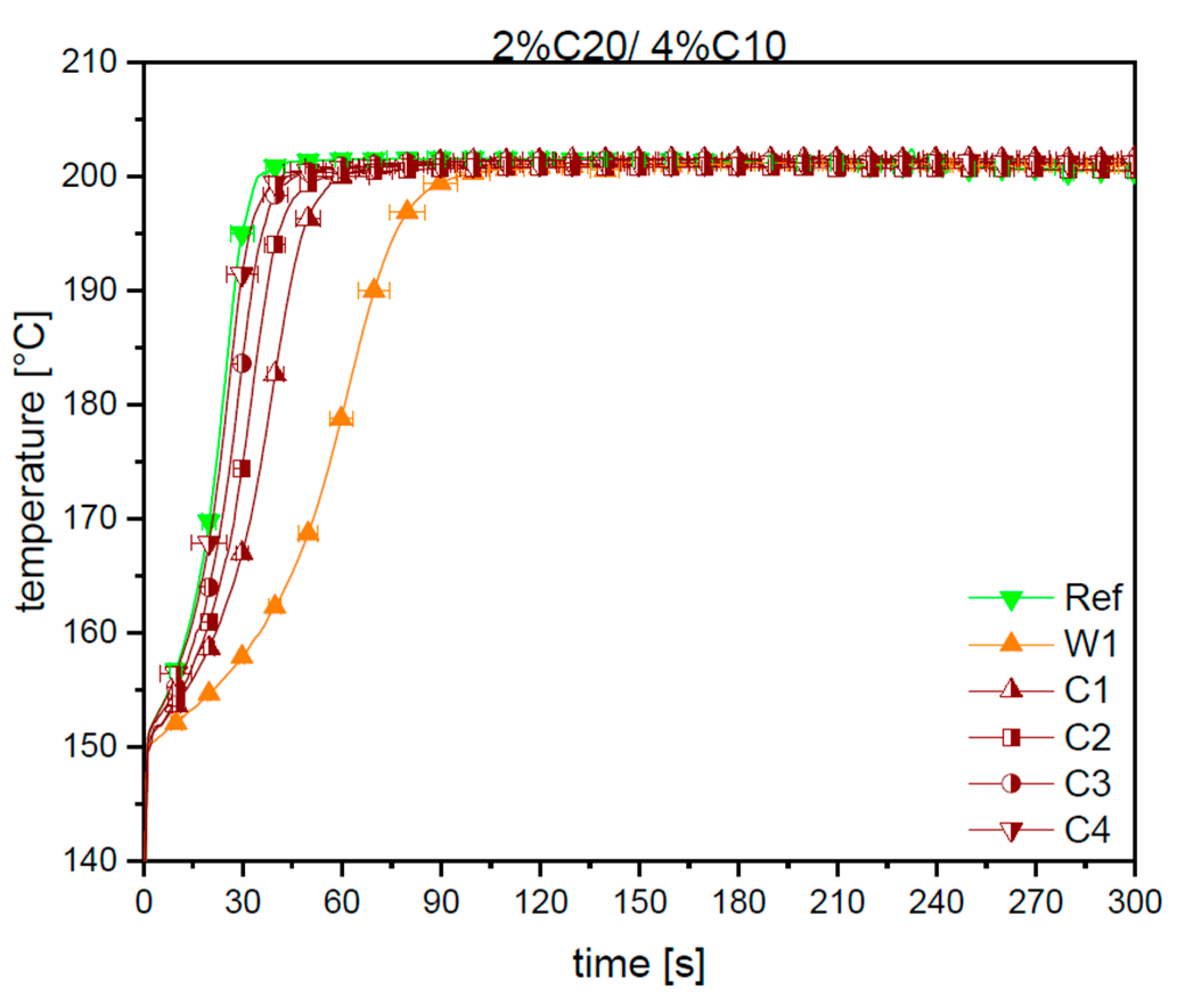
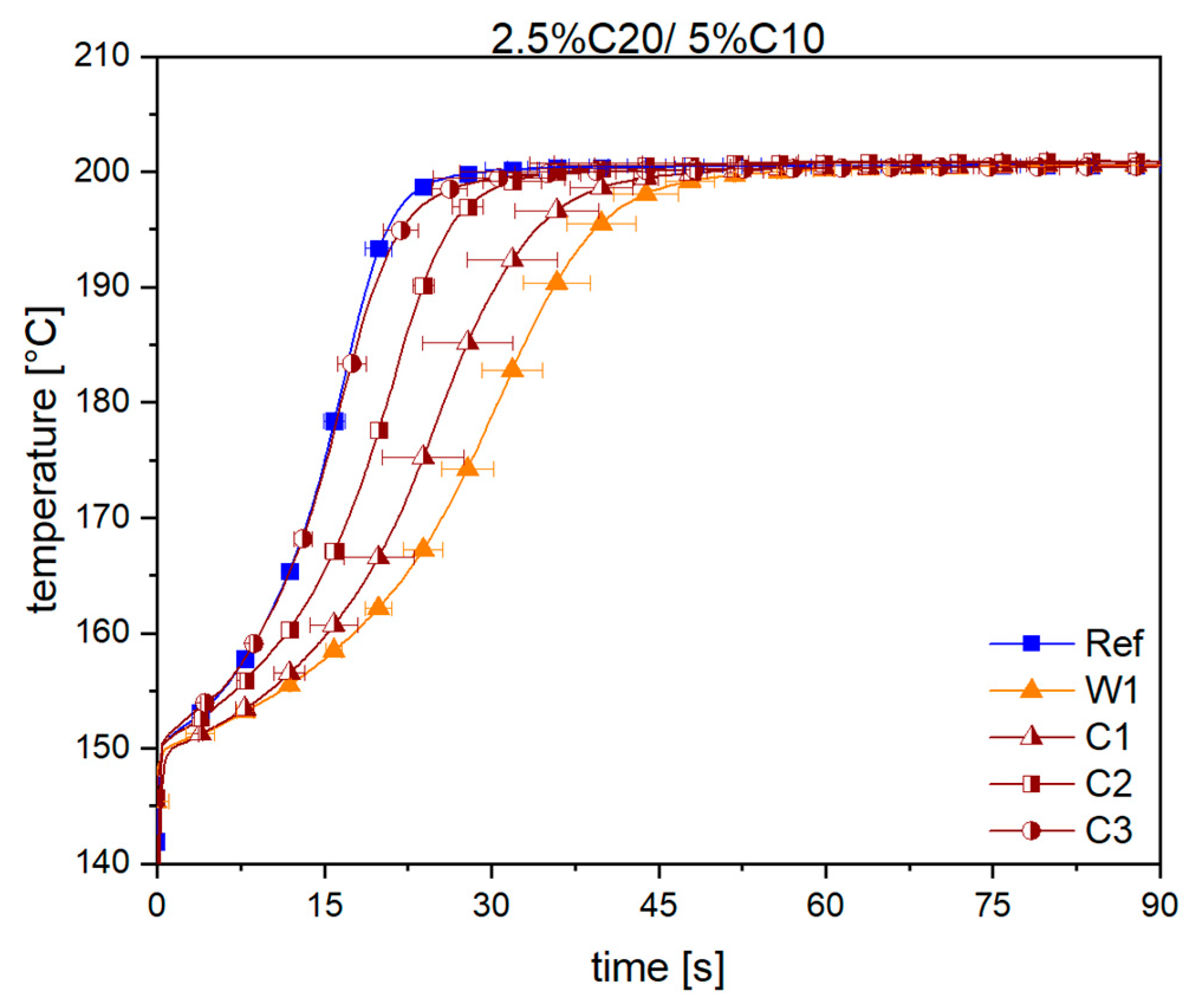
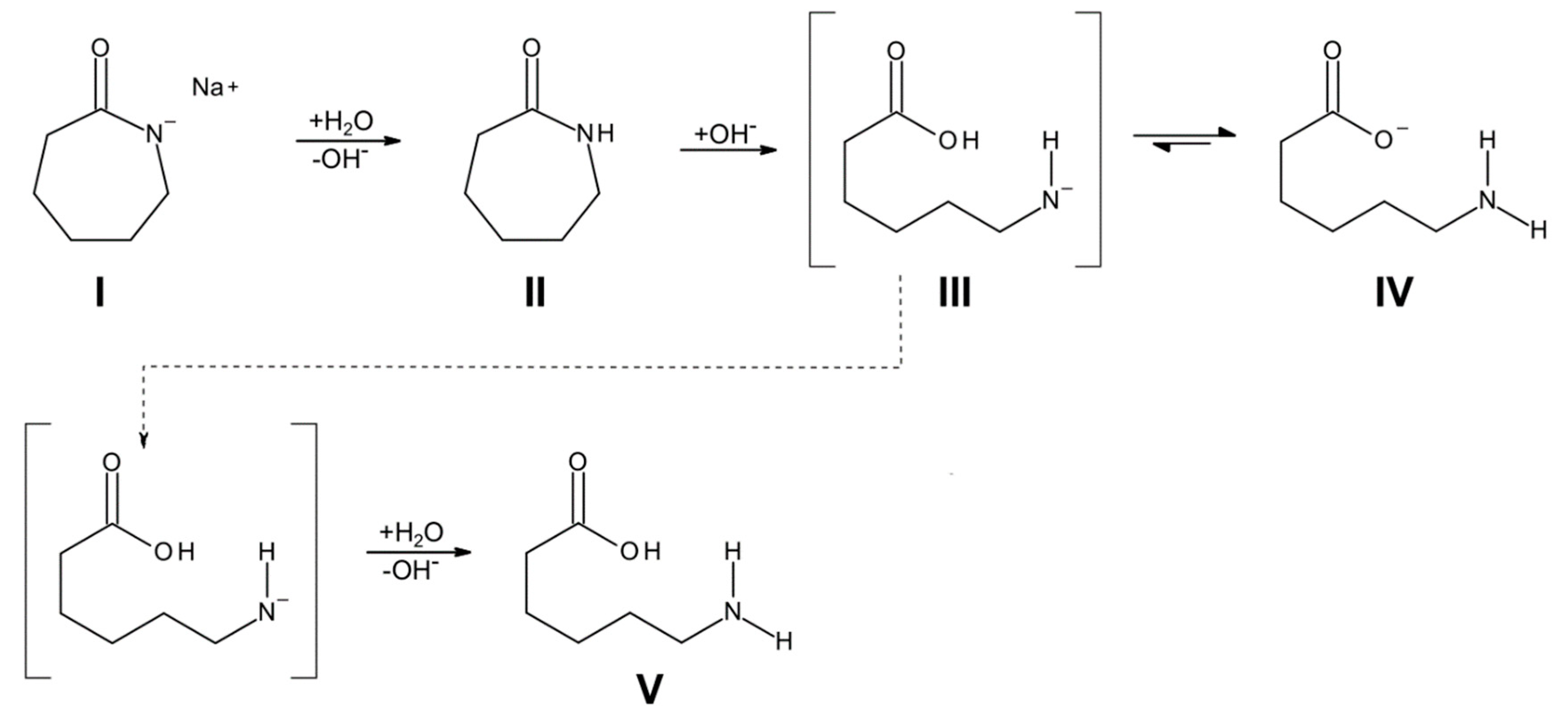
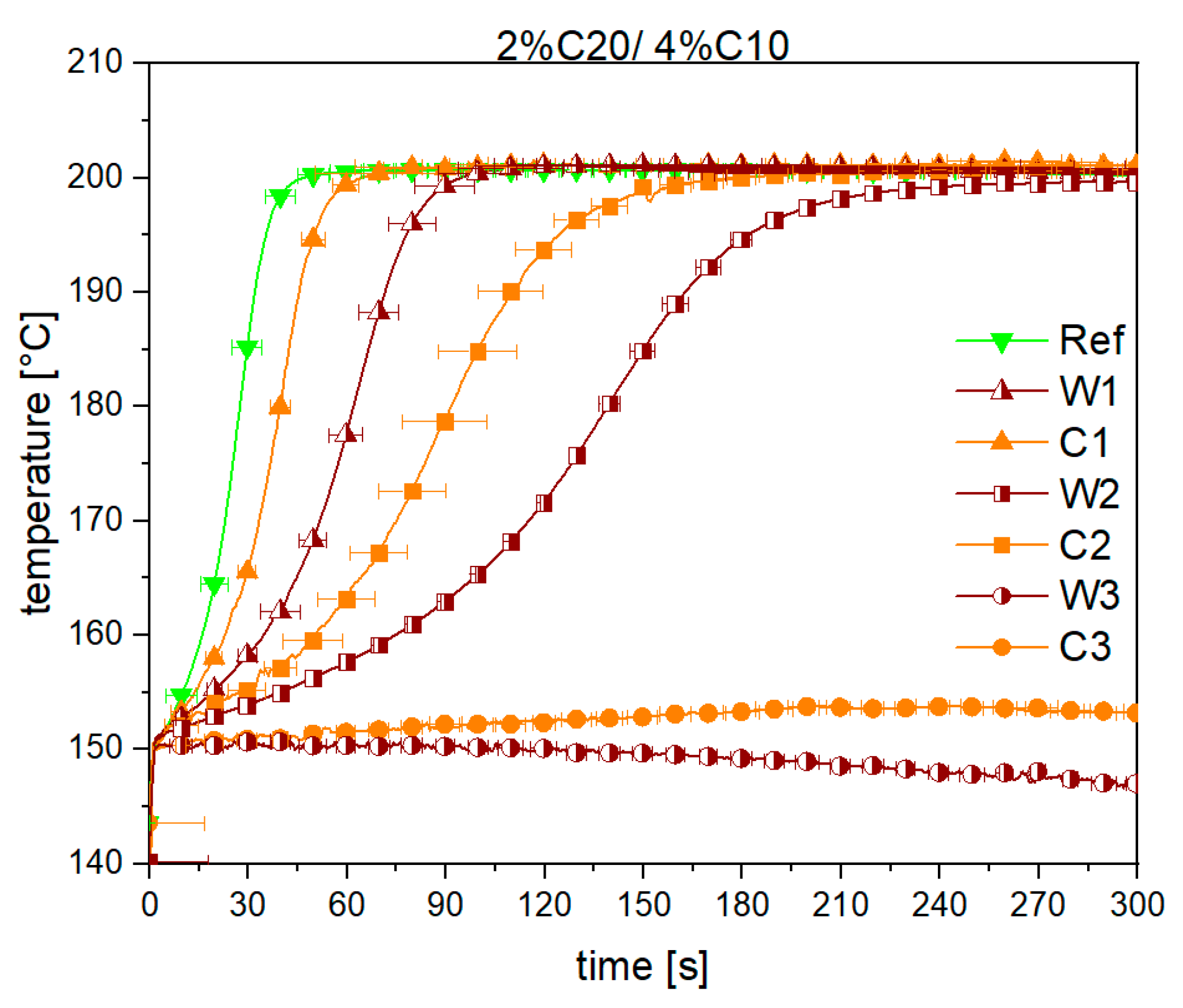
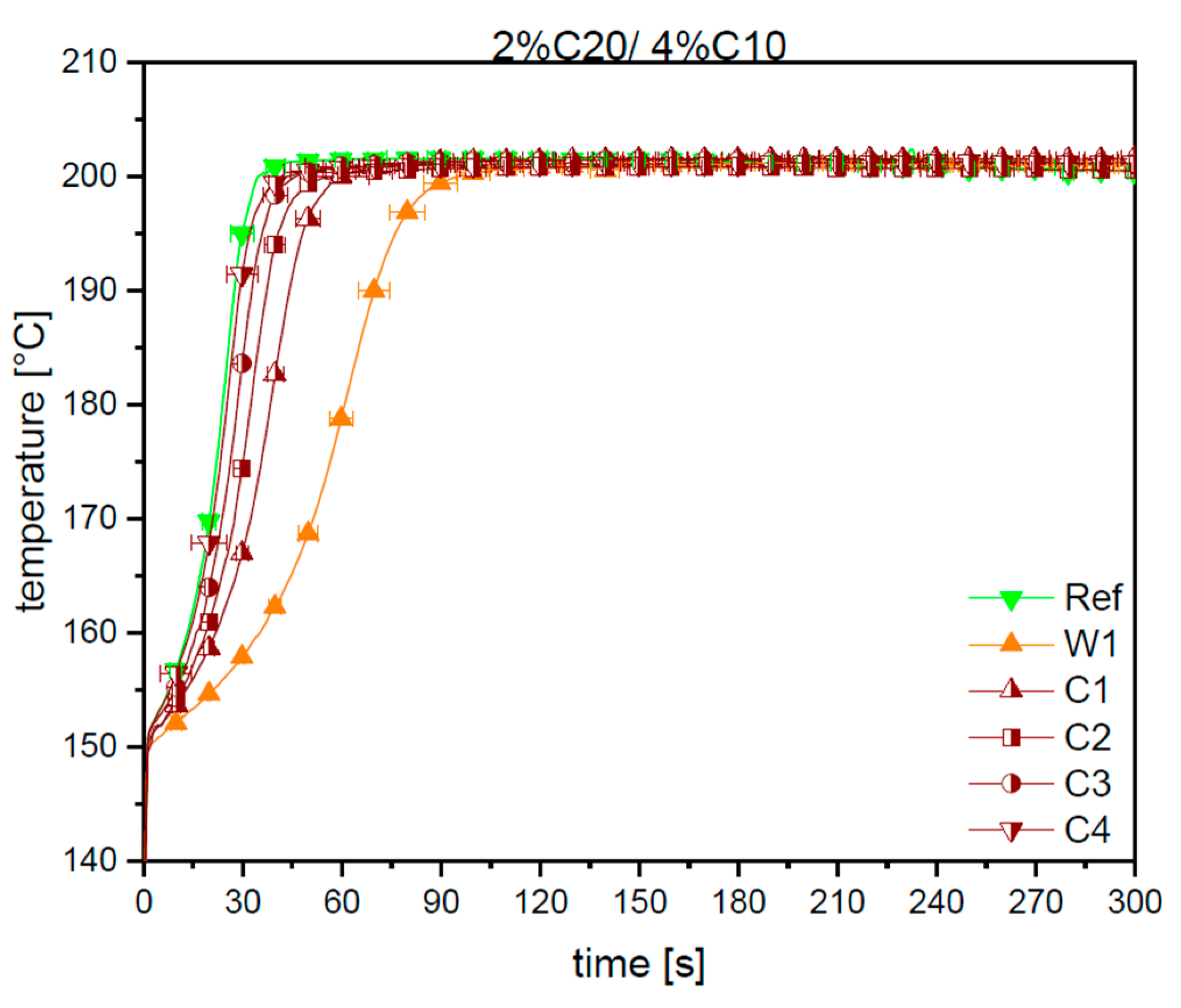
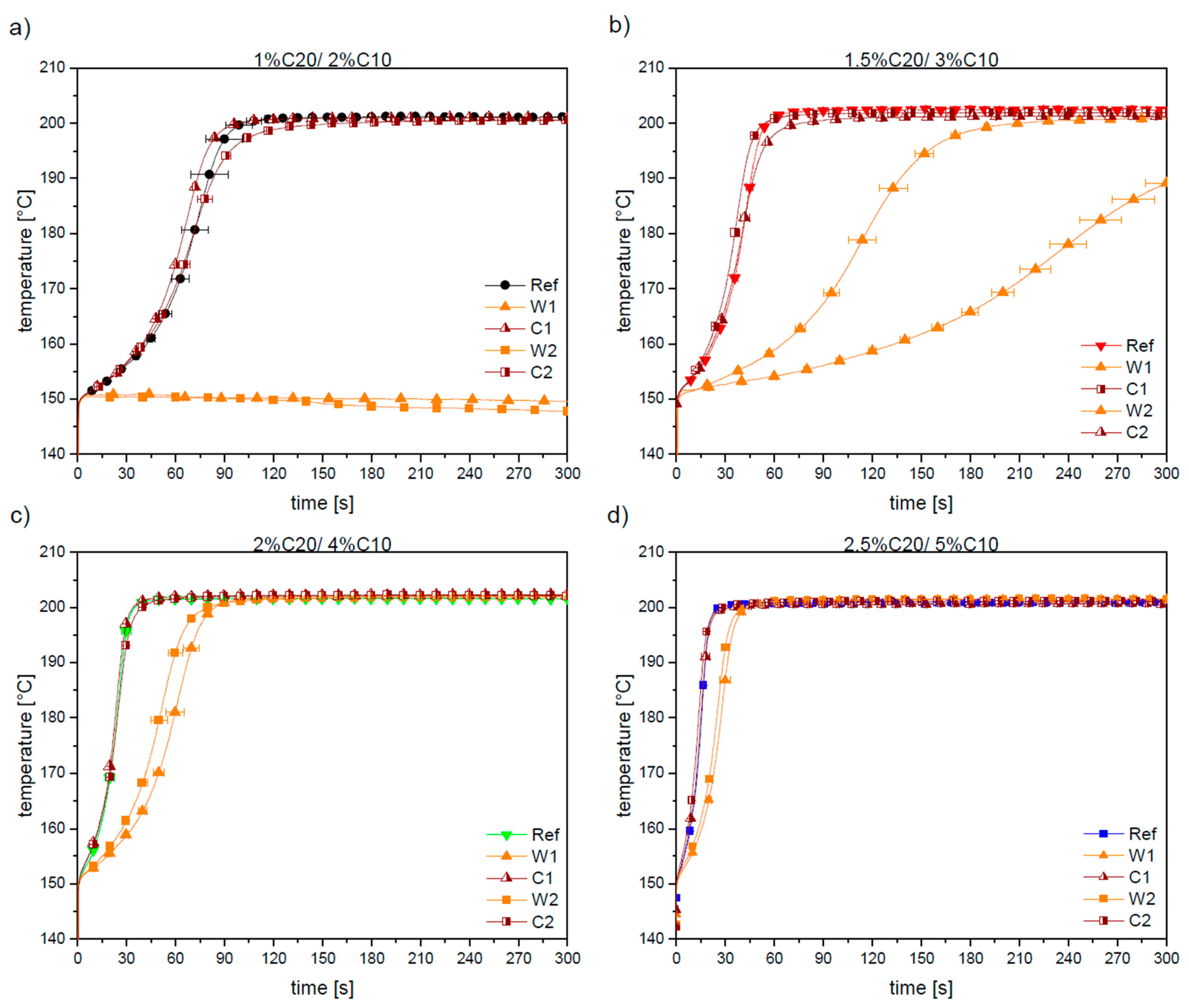
3.1. Reaction Kinetics
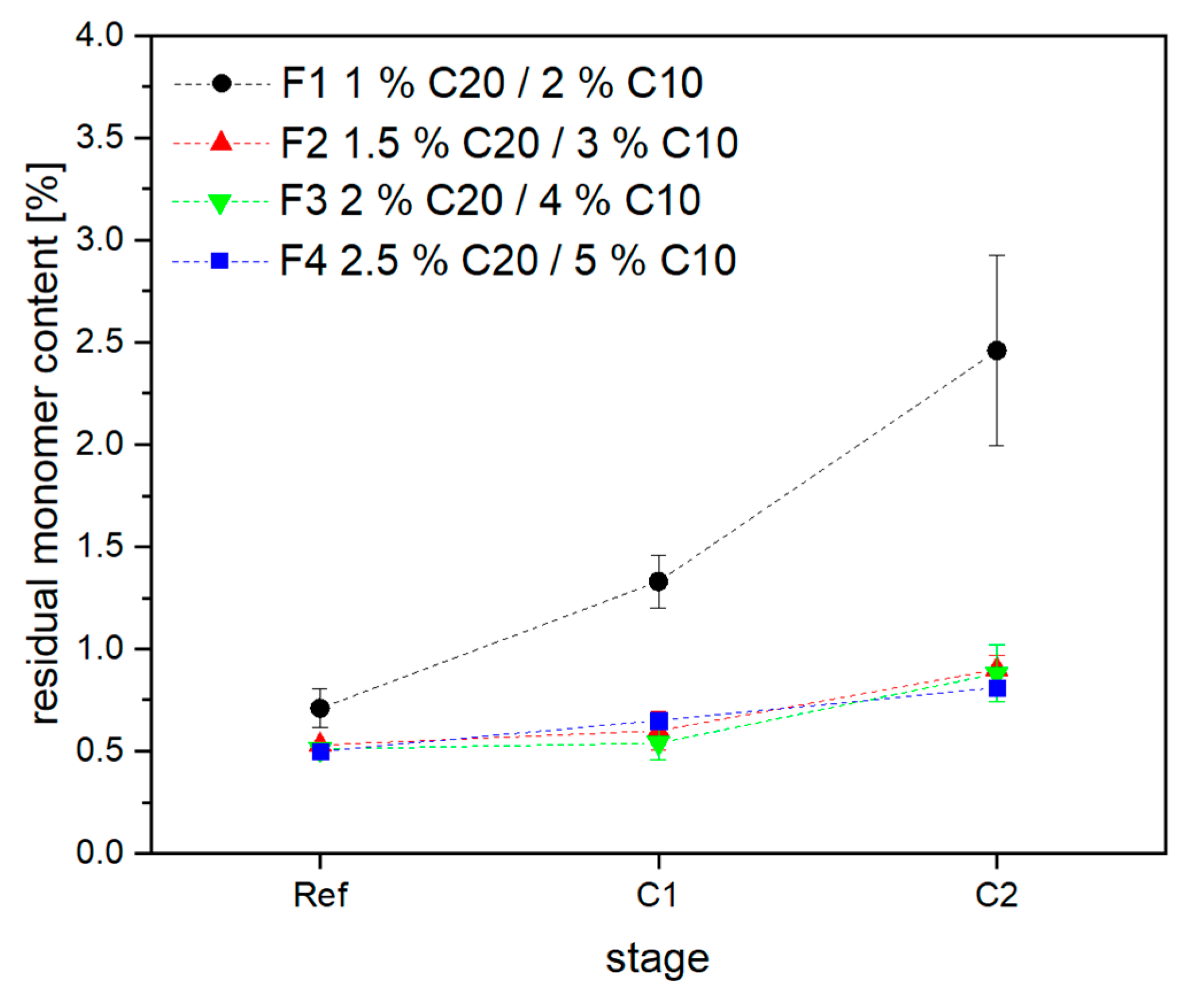
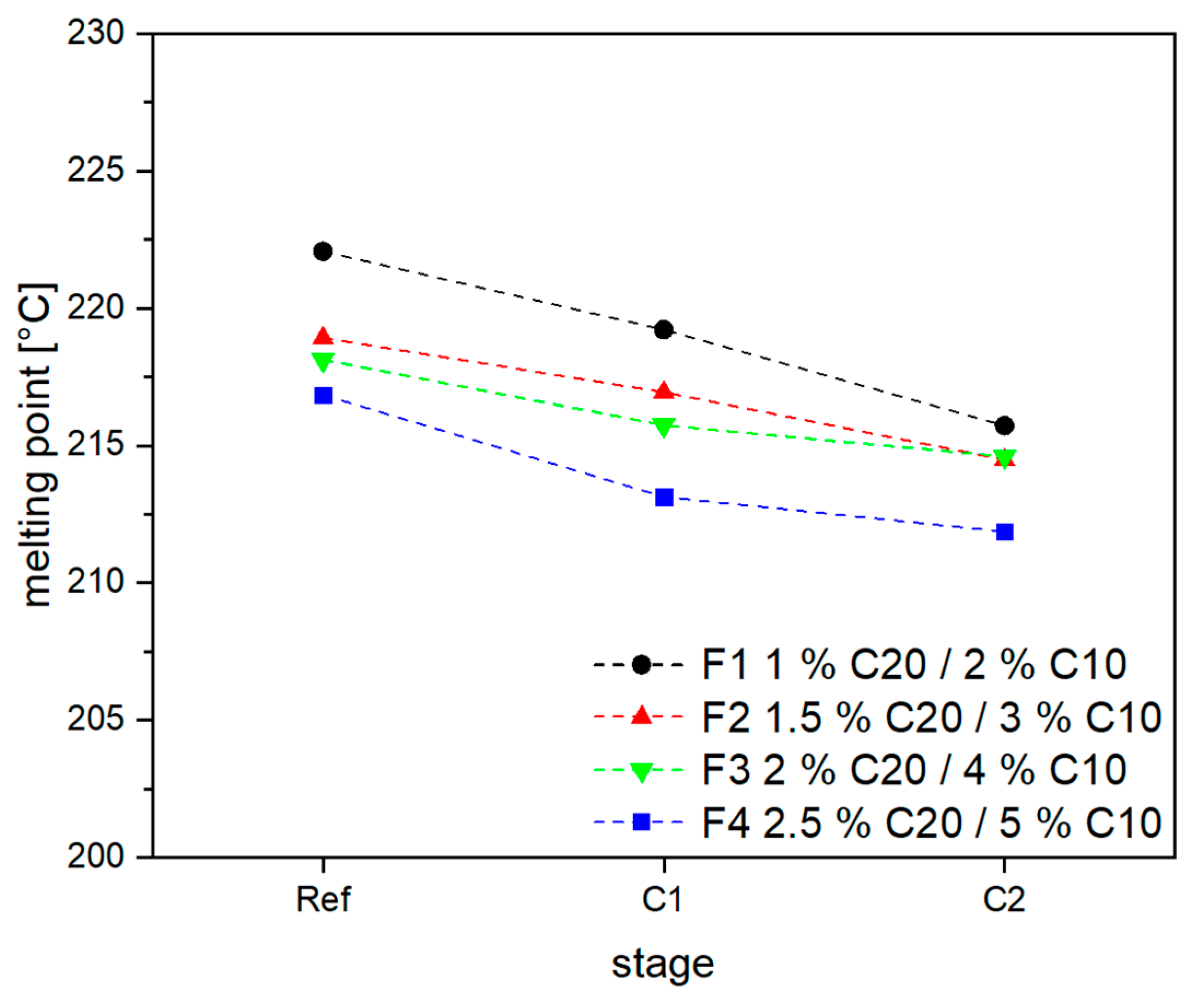
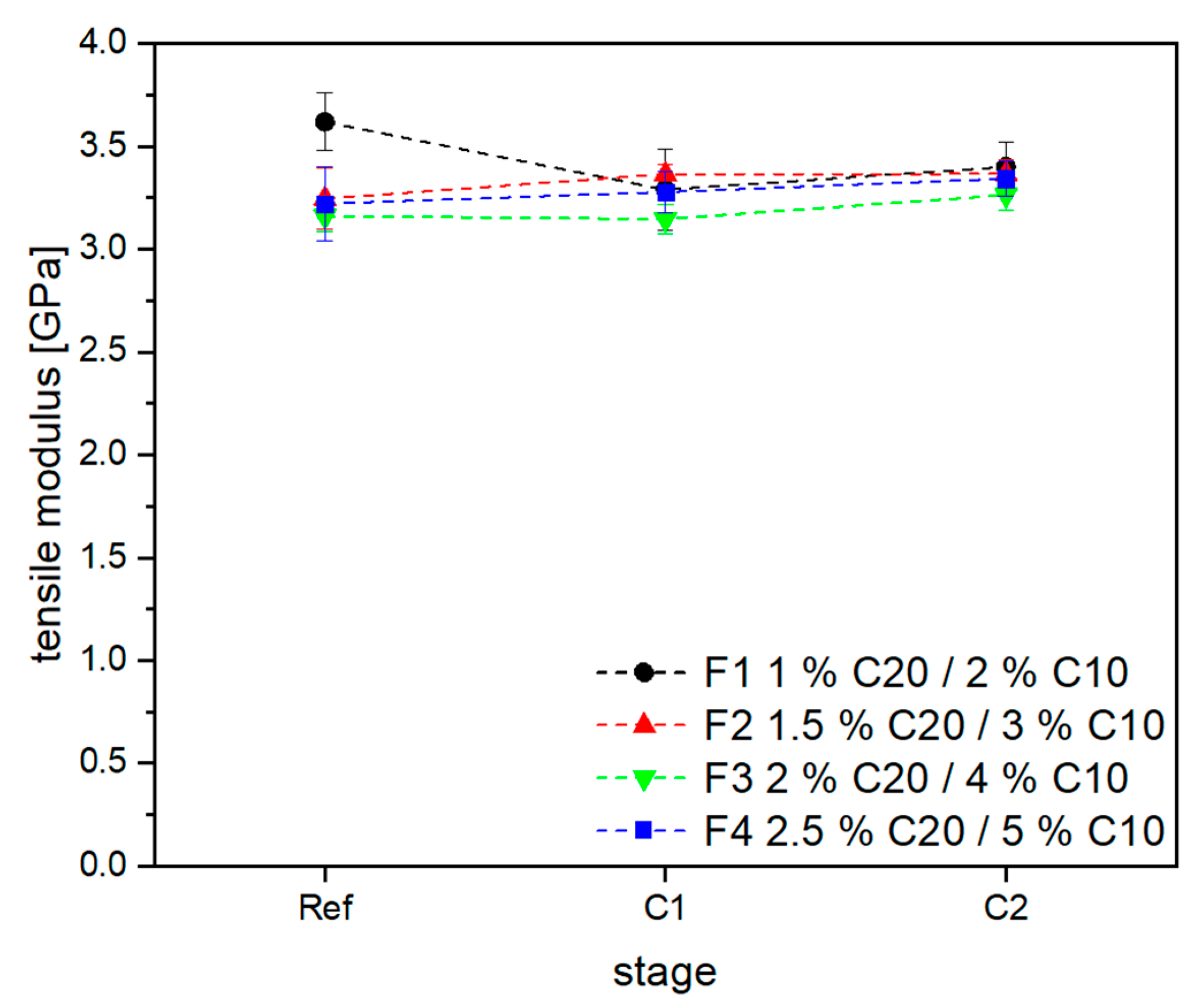
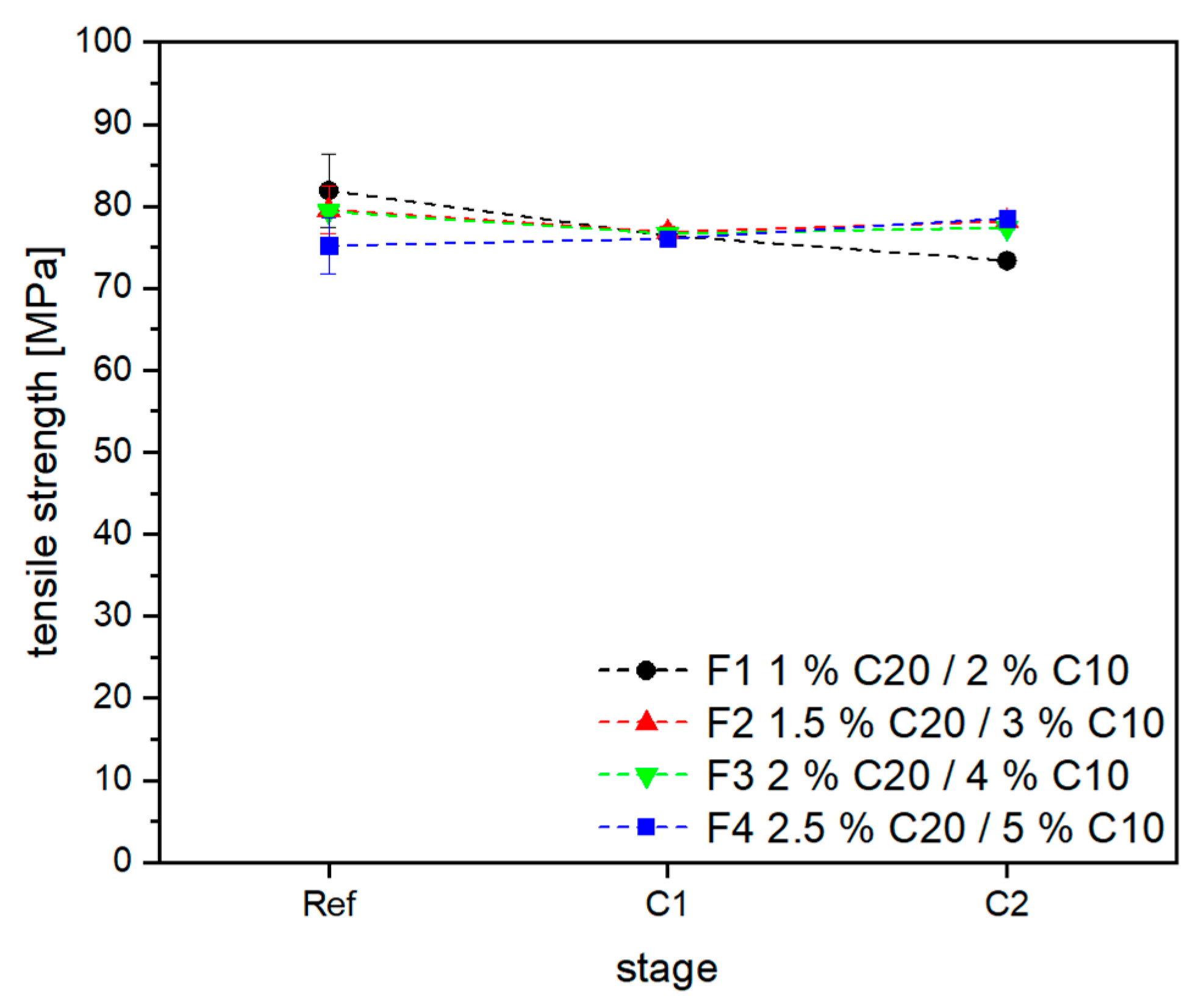
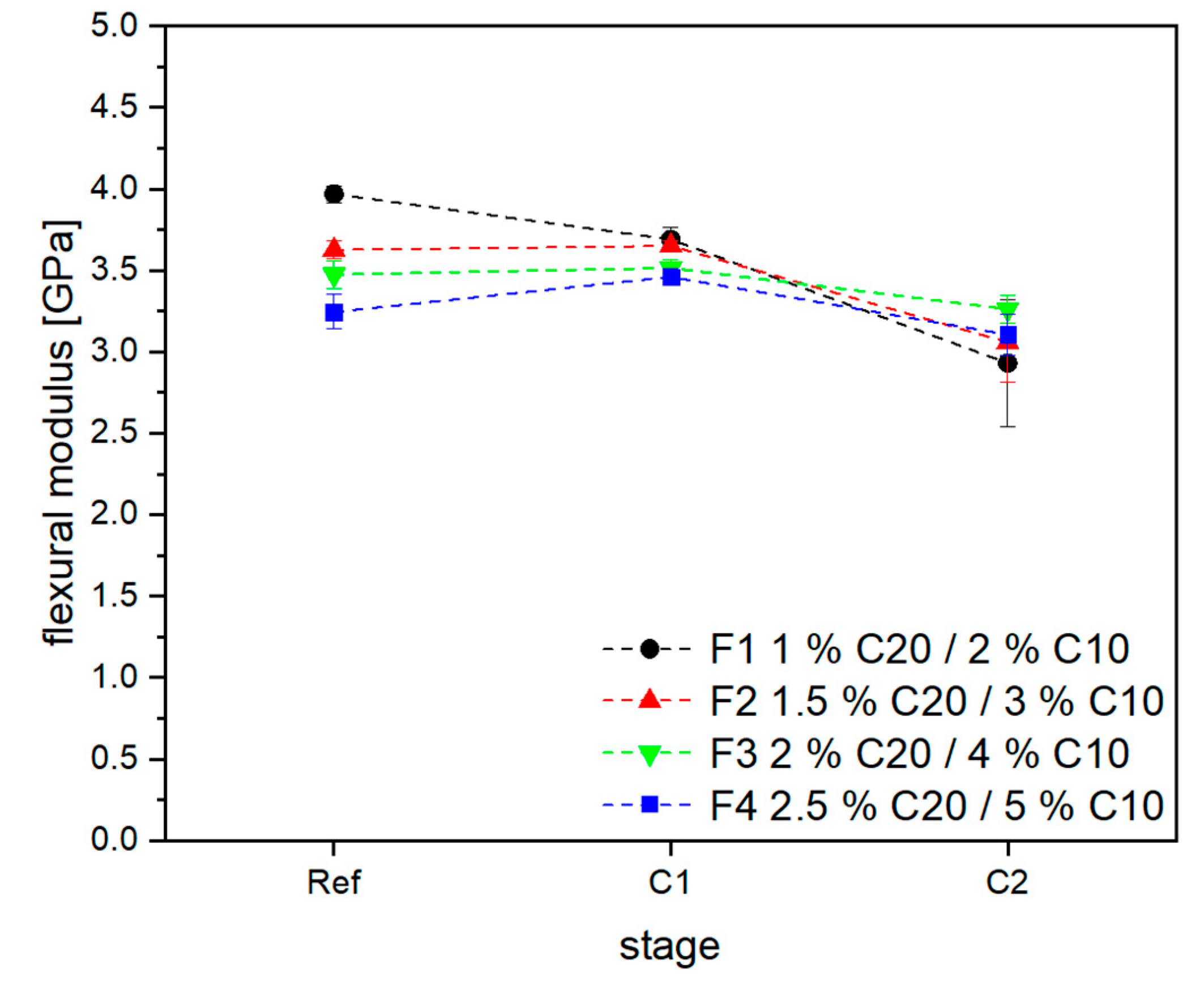
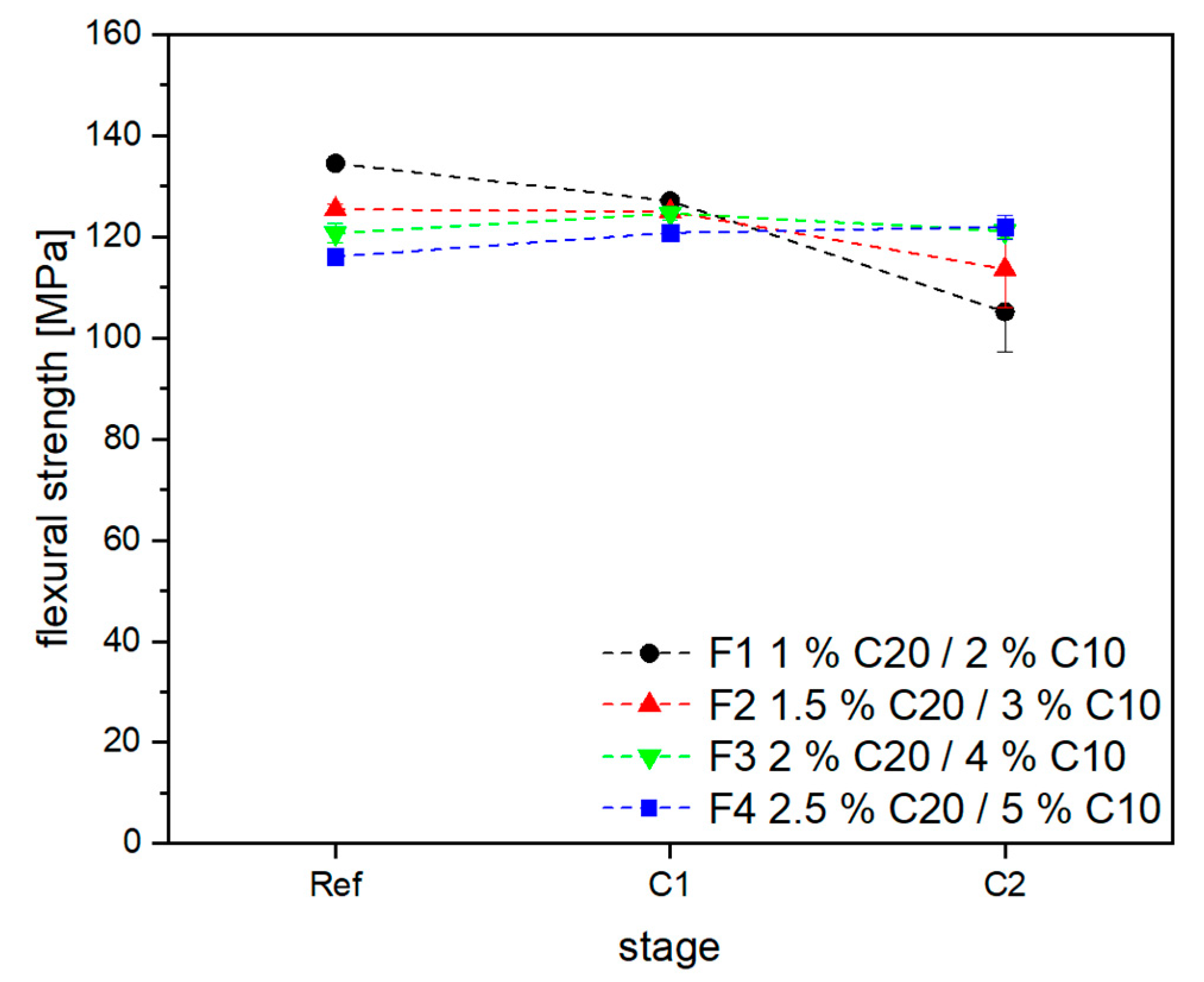
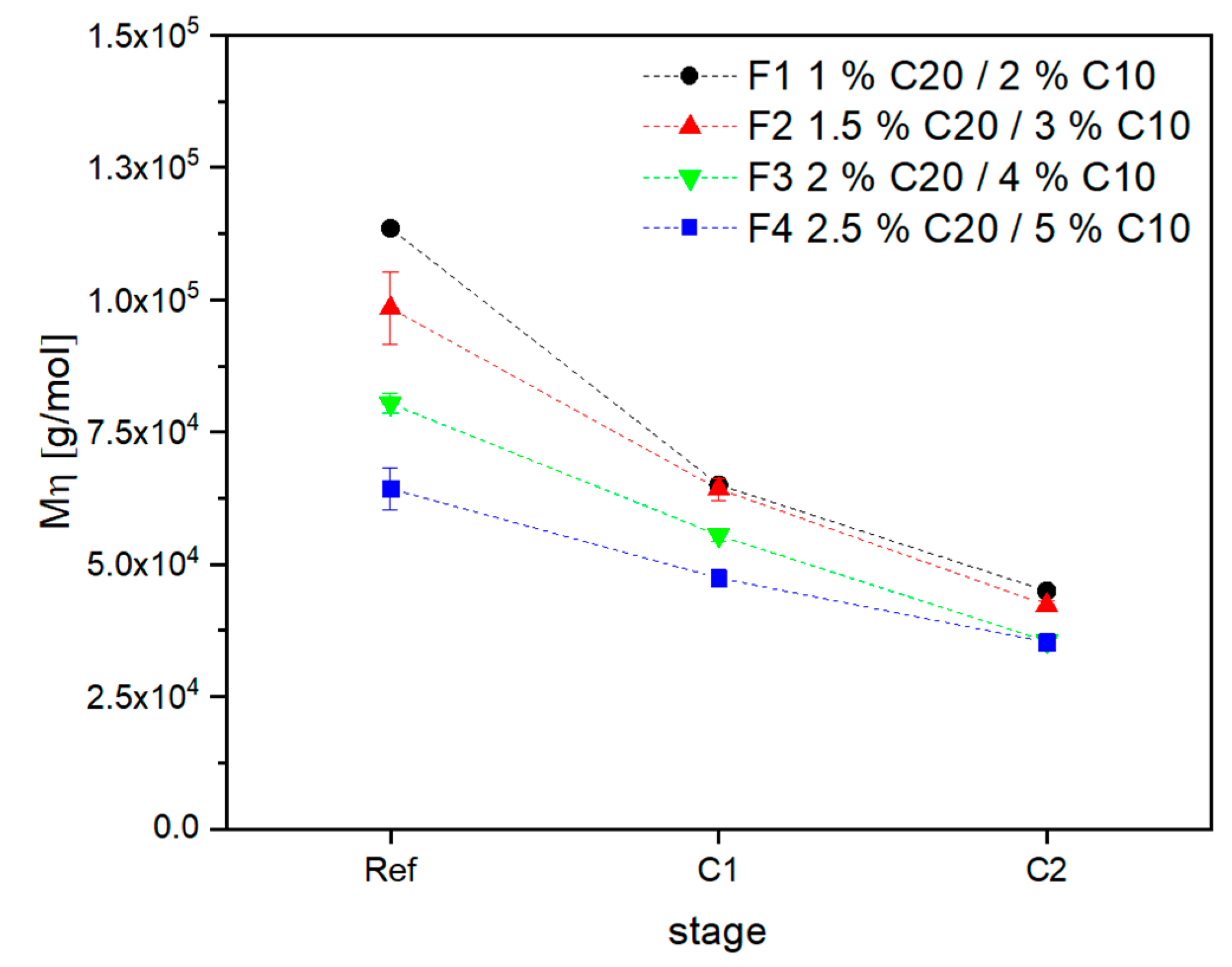
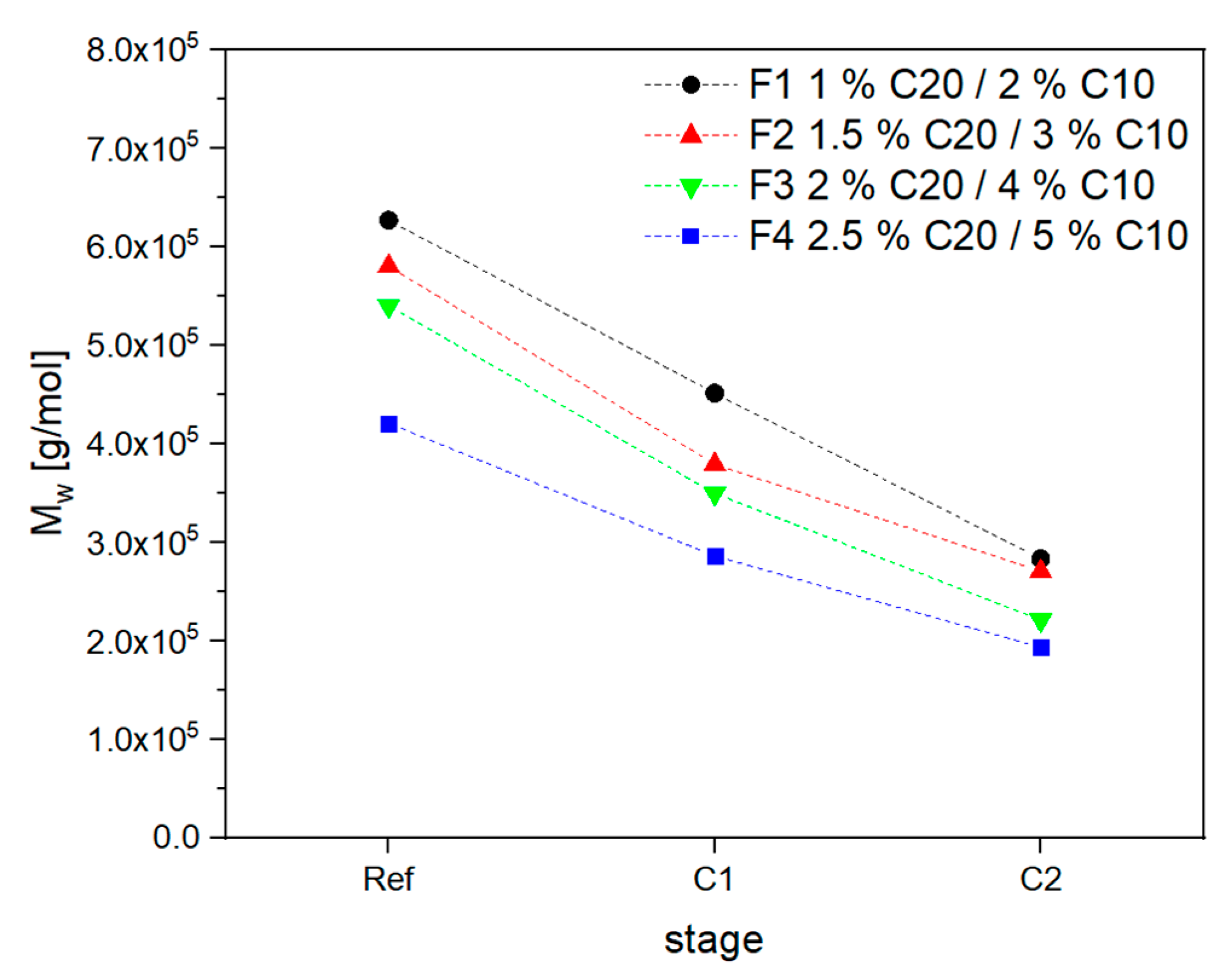
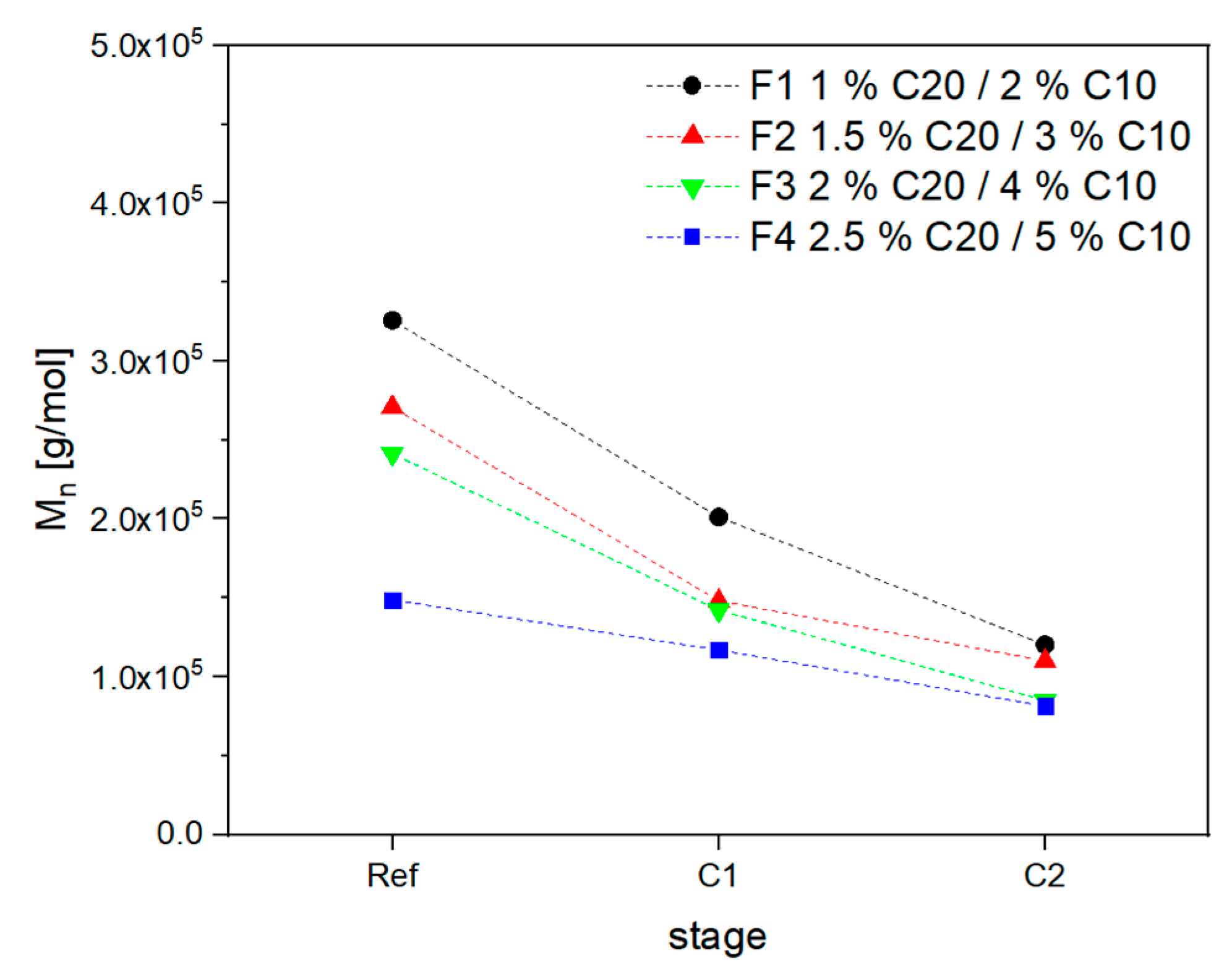
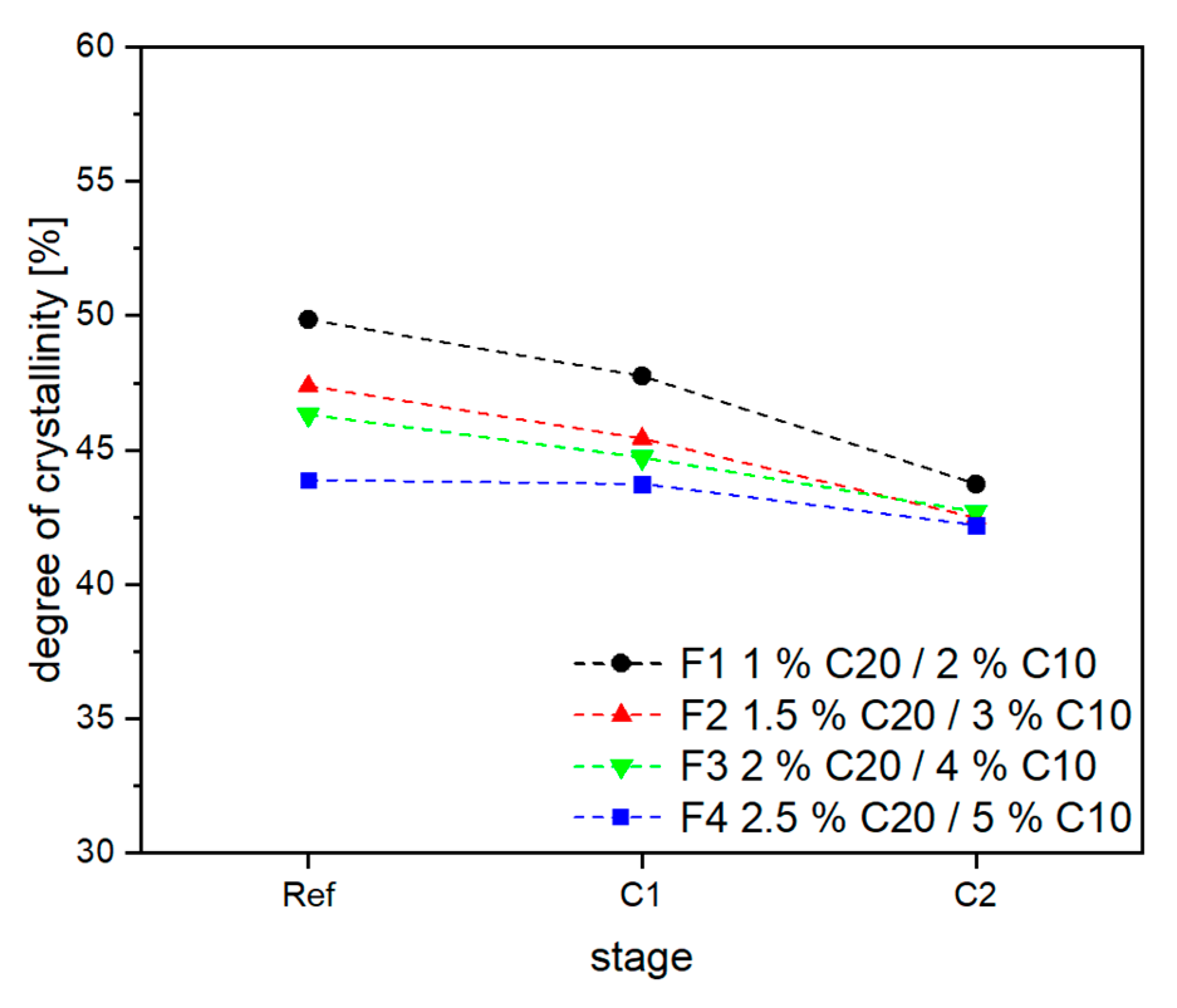
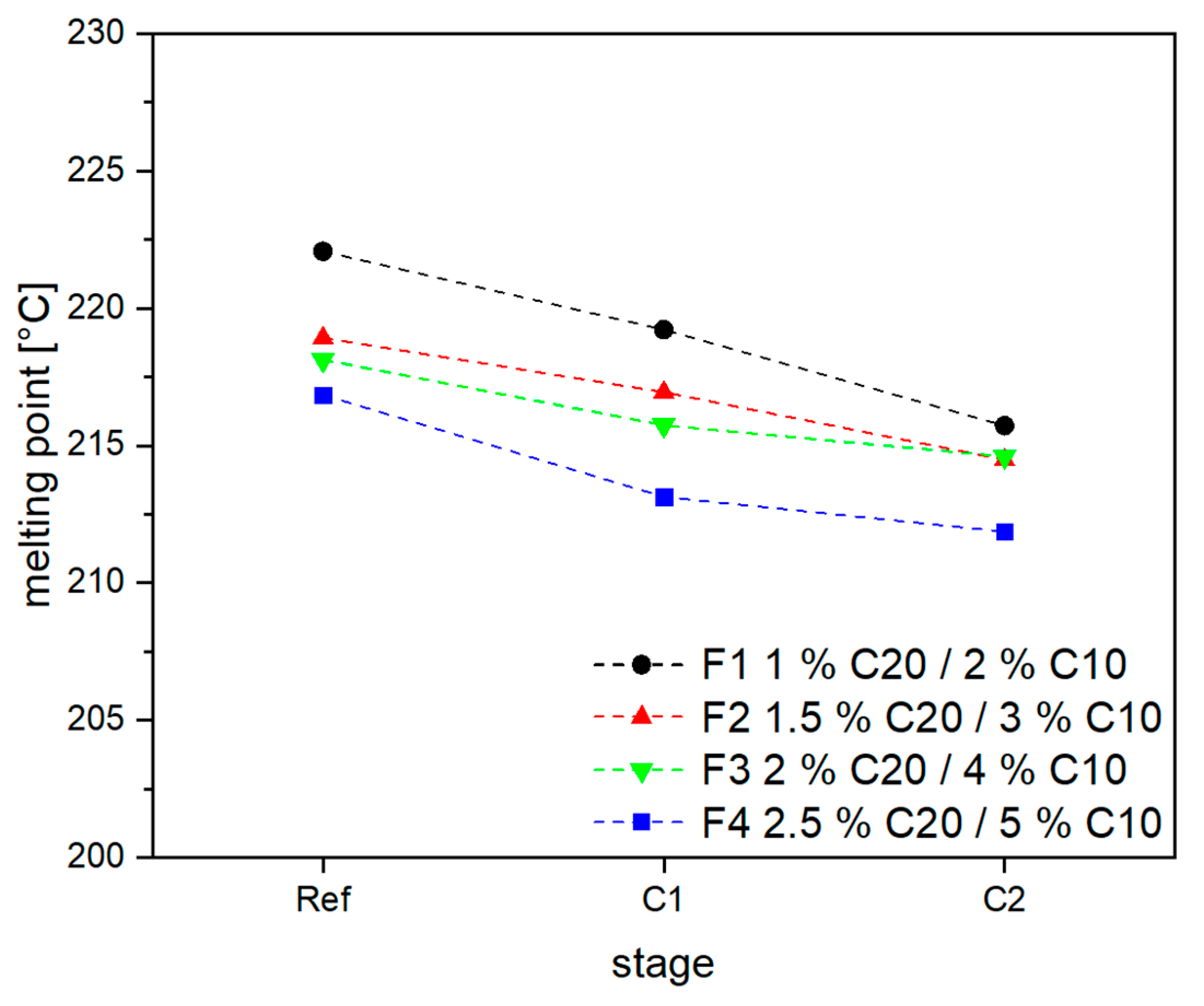
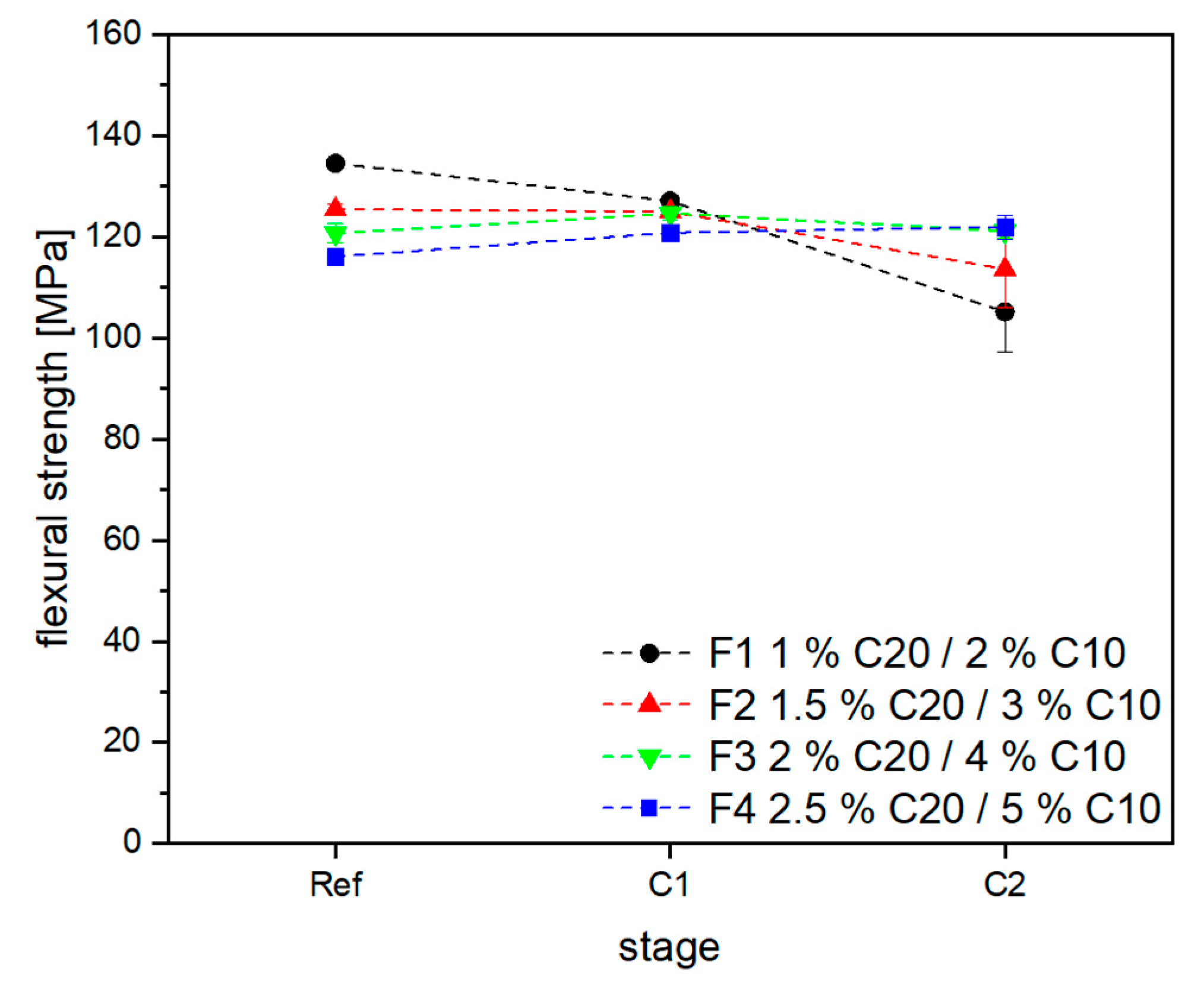
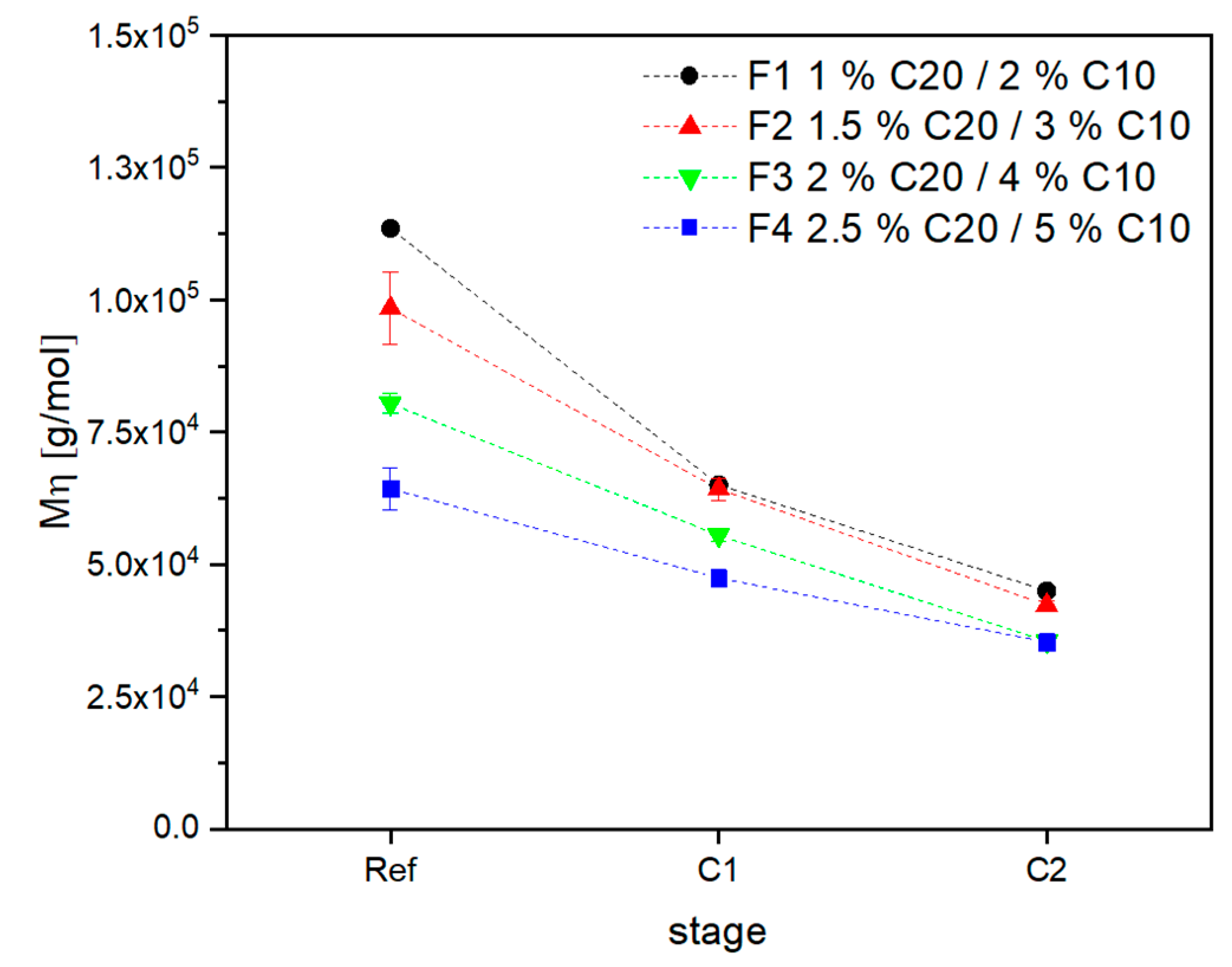
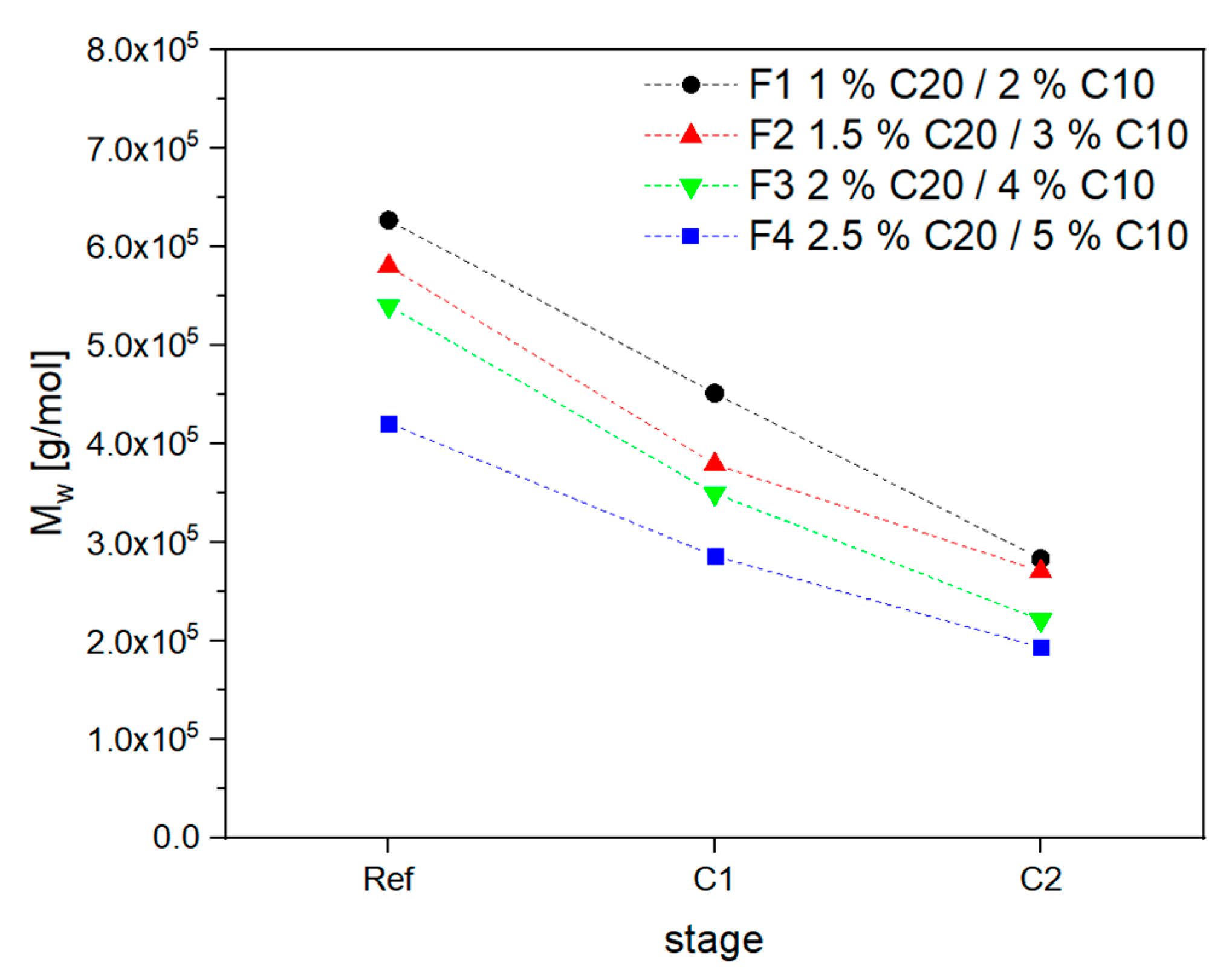
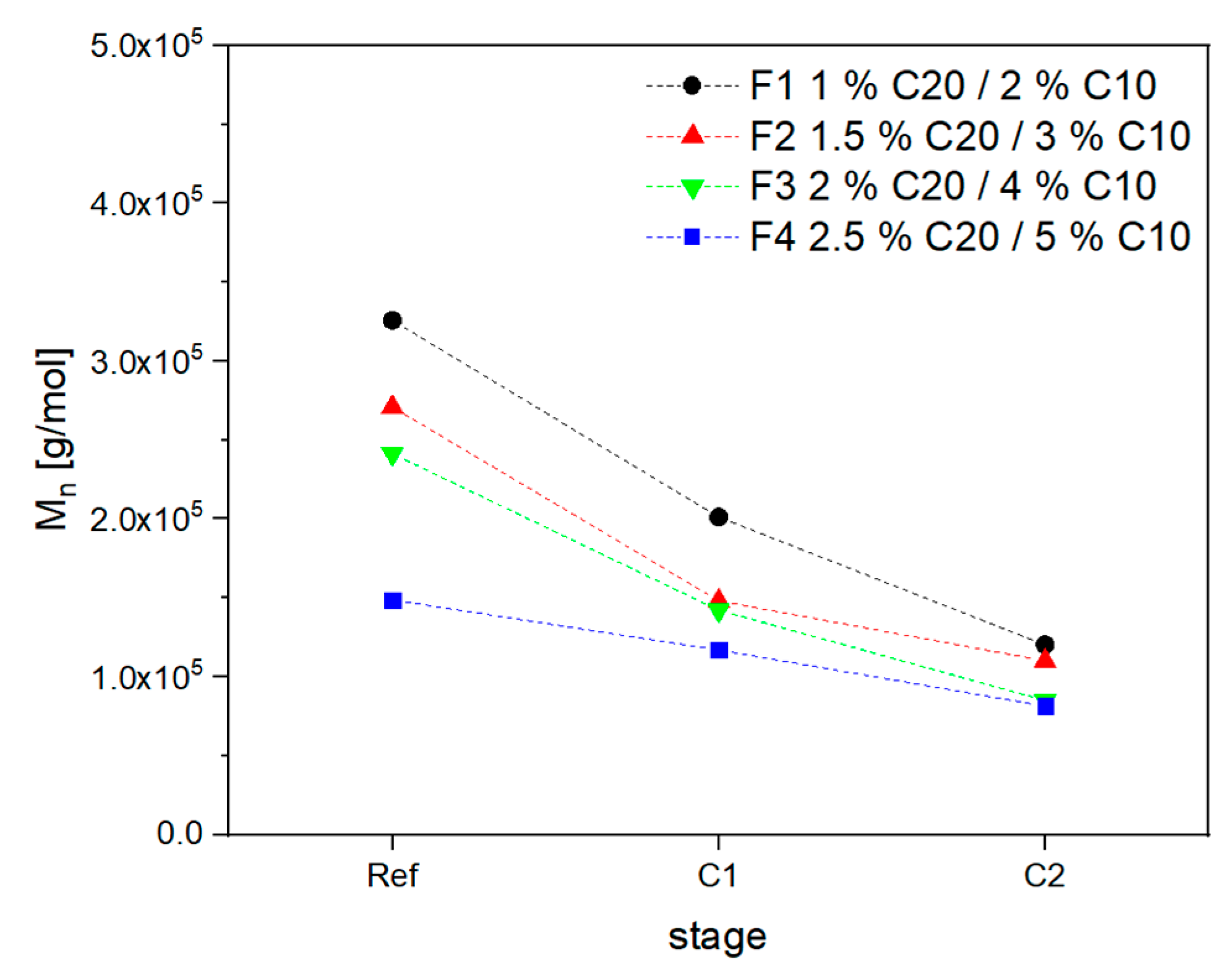
3.2. Material Properties of Compensated APA6 Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Rijswijk, K.; Bersee, H.E.N. Reactive processing of textile fiber-reinforced thermoplastic composites—An overview. Compos. Part. Appl. Sci. Manuf. 2007, 38, 666–681. [Google Scholar] [CrossRef]
- Cho, B.G.; McCarthy, S.P.; Fanucci, J.P.; Nolet, S.C. Fiber reinforced nylon-6 composites produced by the reaction injection pultrusion process. Polym. Compos. 1996, 17, 673–681. [Google Scholar] [CrossRef]
- Garthaus, C.; Witschel, B.; Barfuss, D.; Rohkamm, A.; Gude, M. Funktionalisierte Faser-Thermoplast-Profilstrukturen. Lightweight Des. 2016, 9, 40–45. [Google Scholar] [CrossRef]
- Epple, S.; Bonten, C. Stabile Leichtgewichte-Endlosfaserverstärkte Thermoplasthalbzeuge mit großem Materialquerschnitt. Kunststoffe 2017, 3, 104–106. [Google Scholar]
- Schürmann, H. Konstruieren mit Faser-Kunststoff-Verbunden; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Gittel, D. Gesamtabschlussbericht Verbundprojekt “CastComp”—Neues T-RTM-Verfahren für Hochleistungsverbundstrukturen. 2010. Available online: https://www.tib.eu/de/suchen/?tx_tibsearch_search%5Bdocid%5D=TIBKAT%3A654054894&tx_tibsearch_search%5Bcontroller%5D=Download&cHash=a6f15c07d00109ec4420ff13400163b5#download-mark (accessed on 30 October 2019).
- Maazouz, A.; Lamnawar, K.; Dkier, M. Chemorheological study and in-situ monitoring of PA6 anionic-ring polymerization for RTM processing control. Compos. Part Appl. Sci. Manuf. 2018, 107, 235–247. [Google Scholar] [CrossRef]
- Wendel, R.; Thoma, B.; Henning, F. Influence of water during manufacturing of aPA6 in the thermoplastic RTM process. In Proceedings of the Conference 2017 of the Polymer Processing Society, Cancun, Mexico, 10–14 December 2017; p. 33. [Google Scholar]
- Ageyeva, T.; Sibikin, I.; Karger-Kocsis, J. Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends. Polymers 2018, 10, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- KraussMaffei Technologies GmbH. Misch- und Schmelzvorrichtung zum Schmelzen von schmelzbarem Kunststoffmaterial. DE 102015109770A1, 22 December 2016. [Google Scholar]
- Garcia, A.S.; Palenzuela, G.L. Verfahren und Vorrichtung zur Polymerisation von Lactamen in Formen (US 9290622B2): Google Patents. Available online: https://www.google.com/patents/EP2338665B1?cl=de (accessed on 30 October 2019).
- Bitterlich, M.; Ehleben, M.; Wollny, A.; Desbois, P.; Renkl, J.; Schmidhuber, S. Maßgeschneidert auf reaktives Polyamid 6. Kunststoffe 2014, 104, 80–84. [Google Scholar]
- Fries, E. Heisse Sache—T-RTM-Verfahren und -Demonstrator vereinen Faser, Kunststoff und Metall. Carbon Compos. Mag. 2016, 52, 52–53. [Google Scholar]
- Van Rijswijk, K.; Bersee, H.E.N.; Beukers, A.; Picken, S.J.; Van Geenen, A.A. Optimisation of anionic polyamide-6 for vacuum infusion of thermoplastic composites: Influence of polymerisation temperature on matrix properties. Polym. Test. 2006, 25, 392–404. [Google Scholar] [CrossRef]
- Ueda, K.; Yamada, K.; Nakai, M.; Matsuda, T.; Hosoda, M.; Tai, K. Synthesis of High Molecular Weight Nylon 6 by Anionic Polymerization of ε-Caprolactam. Polym. J. 1996, 28, 446. [Google Scholar] [CrossRef] [Green Version]
- Wittmer, P.; Gerrens, H. Über die anionische Schnellpolymerisation von Caprolactam. Die Makromol. Chem. 1965, 89, 27–43. [Google Scholar] [CrossRef]
- Teuwen, J.J.E.; Van Geenen, A.A.; Bersee, H.E.N. Novel Reaction Kinetic Model for Anionic Polyamide-6. Macromol. Mater. Eng. 2013, 298, 163–173. [Google Scholar] [CrossRef]
- Kim, K.J.; Kim, Y.Y.; Yoon, B.S.; Yoon, K.J. Mechanism and kinetics of adiabatic anionic polymerization of ε-caprolactam in the presence of various activators. J. Appl. Polym. Sci. 1995, 57, 1347–1358. [Google Scholar] [CrossRef]
- Iobst, S.A. Polymerization and crystallization behavior of anionic nylon 6. Polym. Eng. Sci. 1985, 25, 425–430. [Google Scholar] [CrossRef]
- Van Rijswijk, K.; Van Geenen, A.A.; Bersee, H.E.N. Textile fiber-reinforced anionic polyamide-6 composites. Part II: Investigation on interfacial bond formation by short beam shear test. Compos. Part Appl. Sci. Manuf. 2009, 40, 1033–1043. [Google Scholar] [CrossRef]
- Michaeli, W.; Titzschkau, K. BMBF Abschlussbericht: Innovatives Herstellungsverfahren von Polyamid 6-Nanocompounds auf Basis von Schichtsilikaten und ε-Caprolactam. 2009. Available online: https://www.brueggemann.com/de/ap-nylon-additive (accessed on 9 January 2020).
- Elias, H.G. Makromoleküle: Band 3: Industrielle Polymere und Synthesen; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Lechner, M.D.; Gehrke, K.; Nordmeier, E.H. Makromolekulare Chemie: Ein Lehrbuch für Chemiker, Physiker, Materialwissenschaftler und Verfahrenstechniker; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Domininghaus, H.; Elsner, P.; Eyerer, P.; Hirth, T. DOMININGHAUS—Kunststoffe: Eigenschaften und Anwendungen; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Brüggemann Chemical. TDS AP-NYLON Caprolactam flakes. Available online: https://www.brueggemann.com/de/ap-nylon-additive (accessed on 9 January 2020).
- Udipi, K.; Dave, R.S.; Kruse, R.L.; Stebbins, L.R. Polyamides from lactams via anionic ring-opening polymerization: 1. Chemistry and some recent findings. Polymer 1997, 38, 927–938. [Google Scholar] [CrossRef]
- Cartledge, H.C.Y.; Baillie, C.A. Studies of microstructural and mechanical properties of nylon/glass composite Part I The effect of thermal processing on crystallinity, transcrystallinity and crystal phases. J Mater. Sci 1999, 34, 5099–5111. [Google Scholar] [CrossRef]
- Kulicke, W.M.; Clasen, C. Viscosimetry of Polymers and Polyelectrolytes; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Tänzer, W.; Schmidt, S.; Ludwig, I. Molmassebestimmung an Polyamiden und Reaktionsprodukten aus ε-Caprolactam und Phenylglycidether. J. Prakt. Chem. 1996, 338, 311–319. [Google Scholar] [CrossRef]
- Brüggemann Chemical. TDS BRUGGOLEN C10. Available online: https://www.brueggemann.com/de/ap-nylon-additive (accessed on 9 January 2020).
- Brüggemann Chemical. MSDS BRUGGOLEN C20P 2016. Available online: https://www.brueggemann.com/de/ap-nylon-additive (accessed on 9 January 2020).
- Ricco, L.; Russo, S.; Orefice, G.; Riva, F. Anionic Poly(ε-caprolactam): Relationships among Conditions of Synthesis, Chain Regularity, Reticular Order, and Polymorphism. Macromolecules 1999, 32, 7726–7731. [Google Scholar] [CrossRef]
- Weißbach, W.; Dahms, M. Werkstoffkunde: Strukturen, Eigenschaften, Prüfung; Vieweg+Teubner Verlag: Berlin, Germany, 2010. [Google Scholar]
- Dave, R.S.; Kruse, R.L.; Stebbins, L.R.; Udipi, K. Polyamides from lactams via anionic ring-opening polymerization: 2. Kinetics. Polymer 1997, 38, 939–947. [Google Scholar] [CrossRef]
- Malkin, A.Y.L.; Ivanova, S.L.; Frolov, V.G.; Ivanova, A.N.; Andrianova, Z.S. Kinetics of anionic polymerization of lactams. (Solution of non-isothermal kinetic problems by the inverse method). Polymer 1982, 23, 1791–1800. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Theoretical Threshold Value (wt %) | F1: 1% C20/ 2.0% C10 | F2: 1.5% C20/ 3.0% C10 | F3: 2% C20/ 4.0% C10 | F4: 2.5% C20/ 5.0% C10 |
|---|---|---|---|---|
| mH2O, activator | 0.039 | 0.058 | 0.078 | 0.097 |
| mH2O, catalyst | 0.048 | 0.072 | 0.096 | 0.120 |
| mH2O, cor, activator | 0.029 | 0.048 | 0.068 | 0.087 |
| mH2O, cor, catalyst | 0.038 | 0.062 | 0.086 | 0.110 |
| Formulation | Concentration Activator C20P | Concentration Catalyst C10 |
|---|---|---|
| F1 | 1% | 2% |
| F2 | 1.5% | 3% |
| F3 | 2% | 4% |
| F4 | 2.5% | 5% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilhelm, M.; Wendel, R.; Aust, M.; Rosenberg, P.; Henning, F. Compensation of Water Influence on Anionic Polymerization of ε-Caprolactam: 1. Chemistry and Experiments. J. Compos. Sci. 2020, 4, 7. https://doi.org/10.3390/jcs4010007
Wilhelm M, Wendel R, Aust M, Rosenberg P, Henning F. Compensation of Water Influence on Anionic Polymerization of ε-Caprolactam: 1. Chemistry and Experiments. Journal of Composites Science. 2020; 4(1):7. https://doi.org/10.3390/jcs4010007
Chicago/Turabian StyleWilhelm, Michael, Rainer Wendel, Martin Aust, Philipp Rosenberg, and Frank Henning. 2020. "Compensation of Water Influence on Anionic Polymerization of ε-Caprolactam: 1. Chemistry and Experiments" Journal of Composites Science 4, no. 1: 7. https://doi.org/10.3390/jcs4010007
APA StyleWilhelm, M., Wendel, R., Aust, M., Rosenberg, P., & Henning, F. (2020). Compensation of Water Influence on Anionic Polymerization of ε-Caprolactam: 1. Chemistry and Experiments. Journal of Composites Science, 4(1), 7. https://doi.org/10.3390/jcs4010007