Endoplasmic Reticulum Stress in Hearing Loss


Abstract
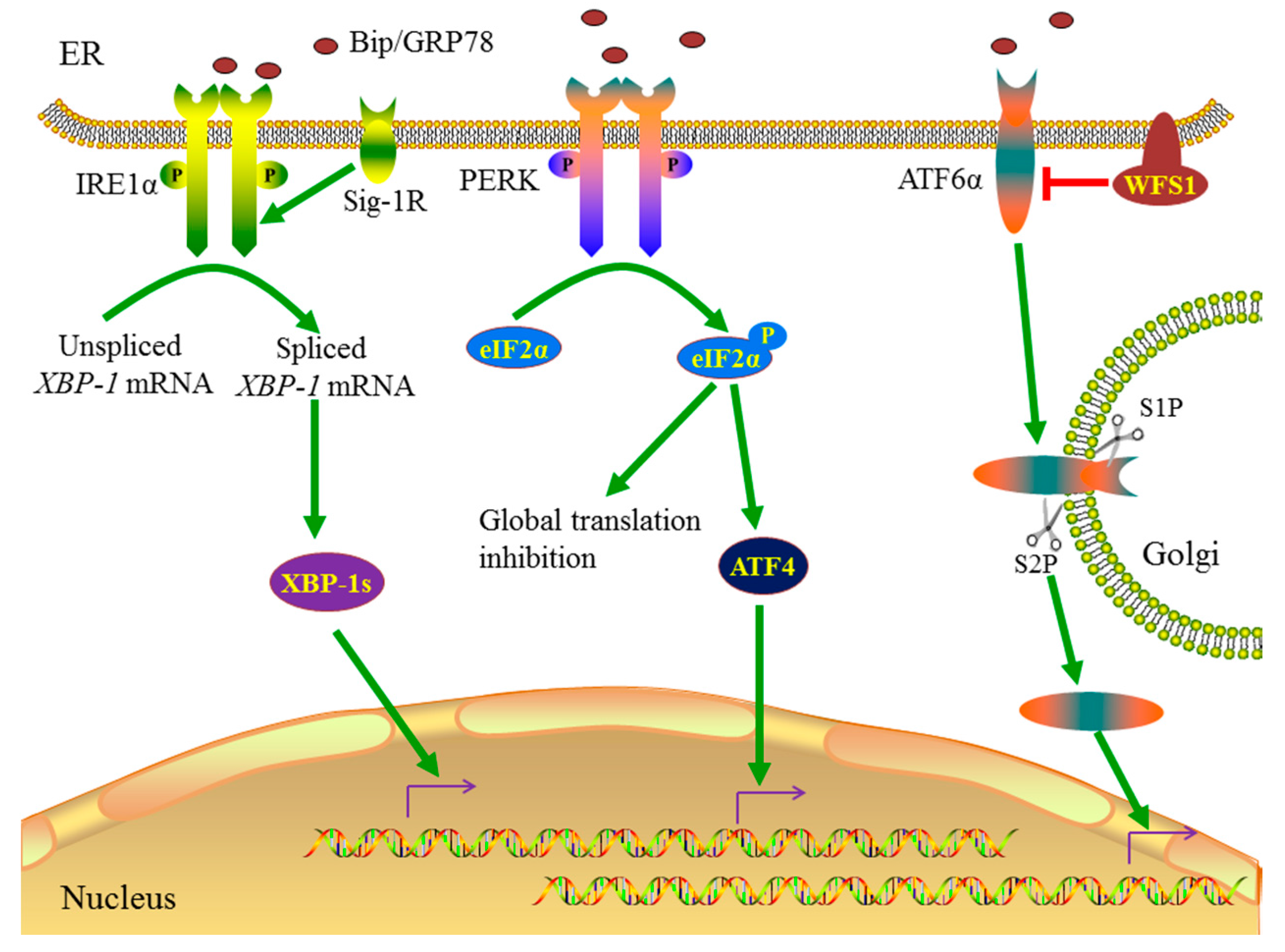
:1. Introduction
2. ER Stress and Drug-Induced Hearing Loss
3. ER Stress and Noise-Induced Hearing Loss
4. ER Stress and Age-Related Hearing Loss
5. ER Stress and Hereditary Hearing Loss
5.1. WFS1 and ER Stress
5.2. Connexins and ER Stress
5.3. USH Proteins and ER Stress
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Bernasconi, R.; Molinari, M. ERAD and ERAD tuning: Disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPS. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BIP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar] [PubMed]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1α is required at 2 distinct steps in b cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The perk eukaryotic initiation factor 2 α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Tsujimura, T.; Takeda, K.; Sugihara, A.; Maekawa, A.; Terada, N.; Yoshida, N.; Akira, S. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells 1998, 3, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hettmann, T.; Barton, K.; Leiden, J.M. Microphthalmia due to p53-mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB-2 transcription factor. Dev. Biol. 2000, 222, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, H.C.; Townes, T.M. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 2002, 99, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Jiao, H.; Li, S.; Cao, H.; Galson, D.L.; Zhao, Z.; Zhao, X.; Lai, Y.; Fan, J.; Im, H.J.; et al. ATF4 promotes bone angiogenesis by increasing vegf expression and release in the bone environment. J. Bone Miner. Res. 2013, 28, 1870–1884. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meng, Q.; Xiao, F.; Chen, S.; Du, Y.; Yu, J.; Wang, C.; Guo, F. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem. J. 2011, 438, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Dyle, M.C.; Kunkel, S.D.; Bullard, S.A.; Bongers, K.S.; Fox, D.K.; Dierdorff, J.M.; Foster, E.D.; Adams, C.M. Stress-induced skeletal muscle GADD45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 2012, 287, 27290–27301. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Zhang, T.; Yu, S.; Lee, S.; Calabuig-Navarro, V.; Yamauchi, J.; Ringquist, S.; Dong, H.H. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J. Biol. Chem. 2013, 288, 25350–25361. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, L.; Selzer, M.E.; Hu, Y. Neuronal endoplasmic reticulum stress in axon injury and neurodegeneration. Ann. Neurol. 2013, 74, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Lanvers-Kaminsky, C.; Zehnhoff-Dinnesen, A.A.; Parfitt, R.; Ciarimboli, G. Drug-induced ototoxicity: Mechanisms, pharmacogenetics, and protective strategies. Clin. Pharmacol. Ther. 2017, 101, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, Y.; Mutai, H.; Kamiya, K.; Mizutari, K.; Fujii, M.; Matsunaga, T. Enhanced expression of C/EBP homologous protein (CHOP) precedes degeneration of fibrocytes in the lateral wall after acute cochlear mitochondrial dysfunction induced by 3-nitropropionic acid. Neurochem. Int. 2010, 56, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Kalinec, G.M.; Thein, P.; Parsa, A.; Yorgason, J.; Luxford, W.; Urrutia, R.; Kalinec, F. Acetaminophen and NAPQI are toxic to auditory cells via oxidative and endoplasmic reticulum stress-dependent pathways. Hear. Res. 2014, 313, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Duscha, S.; Boukari, H.; Meyer, M.; Xie, J.; Wei, G.; Schrepfer, T.; Roschitzki, B.; Boettger, E.C.; Schacht, J. XBP1 mitigates aminoglycoside-induced endoplasmic reticulum stress and neuronal cell death. Cell Death Dis. 2015, 6, e1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinami, Y.; Mutai, H.; Mizutari, K.; Nakagawa, S.; Matsunaga, T. A novel animal model of hearing loss caused by acute endoplasmic reticulum stress in the cochlea. J. Pharmacol. Sci. 2012, 118, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.C. Noise-induced hearing loss: Permanent versus temporary threshold shifts and the effects of hair cell versus neuronal degeneration. Adv. Exp. Med. Biol. 2016, 875, 1–7. [Google Scholar] [PubMed]
- Xue, Q.H.; Chen, J.; Gong, S.S.; He, J.; Xie, J.; Chen, X.L. Role of caspase 12 in apoptosis of cochlea induced by intense noise in guinea pigs. Chin. J. Otorhinolaryngol. Head Neck Surg. 2009, 44, 154–159. [Google Scholar]
- Xue, Q.; Li, C.; Chen, J.; Guo, H.; Li, D.; Wu, X. The protective effect of the endoplasmic reticulum stress-related factors BIP/GRP78 and CHOP/GADD153 on noise-induced hearing loss in guinea pigs. Noise Health 2016, 18, 247–255. [Google Scholar] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the er-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, T.; Omi, T.; Tanimukai, H.; Sakagami, Y.; Tagami, S.; Okochi, M.; Kudo, T.; Takeda, M. Sigma-1Rs are upregulated via PERK/EIF2α/ATF4 pathway and execute protective function in ER stress. Biochem. Biophys. Res. Commun. 2011, 415, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Abdullah, C.S.; Aishwarya, R.; Orr, A.W.; Traylor, J.; Miriyala, S.; Panchatcharam, M.; Pattillo, C.B.; Bhuiyan, M.S. Sigmar1 regulates endoplasmic reticulum stress-induced C/EBP-homologous protein expression in cardiomyocytes. Biosci. Rep. 2017, 37, BSR20170898. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, D.; Sun, G.W.; Cui, Y.; Mita, S.; Otsuki, N.; Kanzaki, S.; Nibu, K.; Ogawa, K.; Matsunaga, T. Neuroprotective effects of cutamesine, a ligand of the sigma-1 receptor chaperone, against noise-induced hearing loss. J. Neurosci. Res. 2015, 93, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, Y.; Chen, S.; Zhou, X.; Wu, X.; Kong, W.; Kong, W. Impaired unfolded protein response in the degeneration of cochlea cells in a mouse model of age-related hearing loss. Exp. Gerontol. 2015, 70, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Hiramatsu, N.; Hayakawa, K.; Okamura, M.; Kasai, A.; Tagawa, Y.; Sawada, N.; Yao, J.; Kitamura, M. Geranylgeranylacetone, an inducer of the 70-kda heat shock protein (HSP70), elicits unfolded protein response and coordinates cellular fate independently ofHSP70. Mol. Pharmacol. 2007, 72, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Hiramatsu, N.; Okamura, M.; Yao, J.; Paton, A.W.; Paton, J.C.; Kitamura, M. Blunted activation of NF-κB and Nf-κB-dependent gene expression by geranylgeranylacetone: Involvement of unfolded protein response. Biochem. Biophys. Res. Commun. 2008, 365, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Mikuriya, T.; Sugahara, K.; Sugimoto, K.; Fujimoto, M.; Takemoto, T.; Hashimoto, M.; Hirose, Y.; Shimogori, H.; Hayashida, N.; Inouye, S.; et al. Attenuation of progressive hearing loss in a model of age-related hearing loss by a heat shock protein inducer, geranylgeranylacetone. Brain Res. 2008, 1212, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Mikuriya, T.; Sugahara, K.; Takemoto, T.; Tanaka, K.; Takeno, K.; Shimogori, H.; Nakai, A.; Yamashita, H. Geranylgeranylacetone, a heat shock protein inducer, prevents acoustic injury in the guinea pig. Brain Res. 2005, 1065, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Song, J.J.; Kim, Y.C.; Park, K.T.; Lee, J.H.; Choi, J.M.; Lee, J.H.; Oh, S.H.; Chang, S.O. Geranylgeranylacetone ameliorates acute cochlear damage caused by 3-nitropropionic acid. Neurotoxicology 2010, 31, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (wolfram syndrome). Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Strom, T.M.; Hortnagel, K.; Hofmann, S.; Gekeler, F.; Scharfe, C.; Rabl, W.; Gerbitz, K.D.; Meitinger, T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (didmoad) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum. Mol. Genet. 1998, 7, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Sanchez, B.; Clement, A.; Fierro, J., Jr.; Washbourne, P.; Westerfield, M. Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Model Mech. 2014, 7, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, B.; Apisa, L.; Yu, H.; Entenman, S.; Xu, M.; Stepanyan, R.; Guan, B.J.; Muller, U.; Hatzoglou, M.; et al. ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23erl/erl mutant mice. Cell Death Dis. 2016, 7, e2485. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (wolfram syndrome 1) gene product: Predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic β-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates er stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Zatyka, M.; Ricketts, C.; da Silva Xavier, G.; Minton, J.; Fenton, S.; Hofmann-Thiel, S.; Rutter, G.A.; Barrett, T.G. Sodium-potassium ATPase 1 subunit is a molecular partner of wolframin, an endoplasmic reticulum protein involved in er stress. Hum. Mol. Genet. 2008, 17, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Yurimoto, S.; Hatano, N.; Tsuchiya, M.; Kato, K.; Fujimoto, T.; Masaki, T.; Kobayashi, R.; Tokumitsu, H. Identification and characterization of wolframin, the product of the wolfram syndrome gene (WFS1), as a novel calmodulin-binding protein. Biochemistry 2009, 48, 3946–3955. [Google Scholar] [CrossRef] [PubMed]
- Gharanei, S.; Zatyka, M.; Astuti, D.; Fenton, J.; Sik, A.; Nagy, Z.; Barrett, T.G. Vacuolar-type H+-ATPase V1a subunit is a molecular partner of wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum. Mol. Genet. 2013, 22, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Philbrook, C.; Gerbitz, K.D.; Bauer, M.F. Wolfram syndrome: Structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum. Mol. Genet. 2003, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Bauer, M.F. Wolfram syndrome-associated mutations lead to instability and proteasomal degradation of wolframin. FEBS Lett. 2006, 580, 4000–4004. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Takeda, S.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Yamada, T.; Inoue, H.; Soga, H.; Katagiri, H.; et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum. Mol. Genet. 2004, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ishihara, H.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Tokita, A.; Satake, C.; Tashiro, F.; Katagiri, H.; et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum. Mol. Genet. 2006, 15, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Bonnet Wersinger, D.; Benkafadar, N.; Jagodzinska, J.; Hamel, C.; Tanizawa, Y.; Lenaers, G.; Delettre, C. Impairment of visual function and retinal er stress activation in WFS1-deficient mice. PLoS ONE 2014, 9, e97222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryns, K.; Thys, S.; Van Laer, L.; Oka, Y.; Pfister, M.; Van Nassauw, L.; Smith, R.J.; Timmermans, J.P.; Van Camp, G. The WFS1 gene, responsible for low frequency sensorineural hearing loss and wolfram syndrome, is expressed in a variety of inner ear cells. Histochem. Cell Biol. 2003, 119, 247–256. [Google Scholar] [PubMed]
- Hansen, L.; Eiberg, H.; Barrett, T.; Bek, T.; Kjaersgaard, P.; Tranebjaerg, L.; Rosenberg, T. Mutation analysis of the WFS1 gene in seven danish wolfram syndrome families; four new mutations identified. Eur. J. Hum. Genet. 2005, 13, 1275–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bespalova, I.N.; Van Camp, G.; Bom, S.J.; Brown, D.J.; Cryns, K.; DeWan, A.T.; Erson, A.E.; Flothmann, K.; Kunst, H.P.; Kurnool, P.; et al. Mutations in the wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum. Mol. Genet. 2001, 10, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Young, T.L.; Ives, E.; Lynch, E.; Person, R.; Snook, S.; MacLaren, L.; Cater, T.; Griffin, A.; Fernandez, B.; Lee, M.K.; et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the wolfram syndrome gene WFS1. Hum. Mol. Genet. 2001, 10, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Cryns, K.; Pfister, M.; Pennings, R.J.; Bom, S.J.; Flothmann, K.; Caethoven, G.; Kremer, H.; Schatteman, I.; Koln, K.A.; Toth, T.; et al. Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum. Genet. 2002, 110, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Bonnycastle, L.L.; Chines, P.S.; Hara, T.; Huyghe, J.R.; Swift, A.J.; Heikinheimo, P.; Mahadevan, J.; Peltonen, S.; Huopio, H.; Nuutila, P.; et al. Autosomal dominant diabetes arising from a wolfram syndrome 1 mutation. Diabetes 2013, 62, 3943–3950. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Tajima, T.; Nakamura, A.; Ishizu, K.; Ariga, T. A novel heterozygous mutation of the WFS1 gene leading to constitutive endoplasmic reticulum stress is the cause of wolfram syndrome. Pediatr. Diabetes 2017. [Google Scholar] [CrossRef] [PubMed]
- De Franco, E.; Flanagan, S.E.; Yagi, T.; Abreu, D.; Mahadevan, J.; Johnson, M.B.; Jones, G.; Acosta, F.; Mulaudzi, M.; Lek, N.; et al. Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes 2017, 66, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- White, T.W.; Bruzzone, R. Multiple connexin proteins in single intercellular channels: Connexin compatibility and functional consequences. J. Bioenerg. Biomembr. 1996, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Willecke, K.; Eiberger, J.; Degen, J.; Eckardt, D.; Romualdi, A.; Guldenagel, M.; Deutsch, U.; Sohl, G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002, 383, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Mila, M.; Monica, M.D.; Lutfi, J.; Shohat, M.; et al. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, F.; Lina-Granade, G.; Plauchu, H.; Bruzzone, R.; Chaib, H.; Levi-Acobas, F.; Weil, D.; Petit, C. Connexin 26 gene linked to a dominant deafness. Nature 1998, 393, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Estivill, X.; Fortina, P.; Surrey, S.; Rabionet, R.; Melchionda, S.; D’Agruma, L.; Mansfield, E.; Rappaport, E.; Govea, N.; Mila, M.; et al. Connexin 26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998, 351, 394–398. [Google Scholar] [CrossRef]
- Kelley, P.M.; Harris, D.J.; Comer, B.C.; Askew, J.W.; Fowler, T.; Smith, S.D.; Kimberling, W.J. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am. J. Hum. Genet. 1998, 62, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, R.; Zelante, L.; Lopez-Bigas, N.; D’Agruma, L.; Melchionda, S.; Restagno, G.; Arbones, M.L.; Gasparini, P.; Estivill, X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 2000, 106, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Grifa, A.; Wagner, C.A.; D’Ambrosio, L.; Melchionda, S.; Bernardi, F.; Lopez-Bigas, N.; Rabionet, R.; Arbones, M.; Monica, M.D.; Estivill, X.; et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 1999, 23, 16–18. [Google Scholar] [PubMed]
- Del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Telleria, D.; Menendez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Z.; Xia, X.J.; Xu, L.R.; Pandya, A.; Liang, C.Y.; Blanton, S.H.; Brown, S.D.; Steel, K.P.; Nance, W.E. Mutations in connexin31 underlie recessive as well as dominant non-syndromic hearing loss. Hum. Mol. Genet. 2000, 9, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Huang, S.H.; Chou, K.H.; Liao, P.J.; Su, C.C.; Li, S.Y. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in taiwan. Audiol. Neuro-Otol. 2007, 12, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wan, Y.; Zhu, Y.; Fang, X.; Hiramatsu, N.; Hayakawa, K.; Paton, A.W.; Paton, J.C.; Kitamura, M.; Yao, J. Downregulation of gap junction expression and function by endoplasmic reticulum stress. J. Cell. Biochem. 2009, 107, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Klee, P.; Cardozo, A.K.; Meda, P.; Haefliger, J.A. Connexin36 contributes to INS-1E cells survival through modulation of cytokine-induced oxidative stress, ER stress and AMPK activity. Cell Death Differ. 2013, 20, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, D.; Scott, C.A.; Gray, C.; Zicha, D.; Kelsell, D.P. EKV mutant connexin 31 associated cell death is mediated by ER stress. Hum. Mol. Genet. 2009, 18, 4734–4745. [Google Scholar] [CrossRef] [PubMed]
- Alapure, B.V.; Stull, J.K.; Firtina, Z.; Duncan, M.K. The unfolded protein response is activated in connexin 50 mutant mouse lenses. Exp. Eye Res. 2012, 102, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Minogue, P.J.; Lambert, P.A.; Snabb, J.I.; Beyer, E.C. The cataract-linked mutant connexin50D47A causes endoplasmic reticulum stress in mouse lenses. J. Biol. Chem. 2016, 291, 17569–17578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Li, L.; Sun, Y.; Feng, B. Three common GJB2 mutations causing nonsyndromic hearing loss in Chinese populations are retained in the endoplasmic reticulum. Acta Oto-Laryngol. 2010, 130, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.M.; Yang, J.J.; Su, C.C.; Chang, J.Y.; Li, T.C.; Li, S.Y. A novel mutation in the connexin 29 gene may contribute to nonsyndromic hearing loss. Hum. Genet. 2010, 127, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Wang, W.H.; Chen, P.H.; Li, S.Y.; Yang, J.J. Functional analysis of a nonsyndromic hearing loss-associated mutation in the transmembrane II domain of the GJC3 gene. Int. J. Med. Sci. 2017, 14, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Ma, H.; Xiong, H.; Pan, Q.; Huang, L.; Wang, D.; Zhang, Z. Trafficking abnormality and ER stress underlie functional deficiency of hearing impairment-associated connexin-31 mutants. Protein Cell 2010, 1, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Kelly, J.J.; Lajoie, P.; Shao, Q.; Laird, D.W. Mutations in CX30 that are linked to skin disease and non-syndromic hearing loss exhibit several distinct cellular pathologies. J. Cell Sci. 2014, 127, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Boughman, J.A.; Vernon, M.; Shaver, K.A. Usher syndrome: Definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 1983, 36, 595–603. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Isosomppi, J.; Vastinsalo, H.; Geller, S.F.; Heon, E.; Flannery, J.G.; Sankila, E.M. Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane. Mol. Vis. 2009, 15, 1806–1818. [Google Scholar] [PubMed]
- Han, F.; Yu, H.; Tian, C.; Chen, H.E.; Benedict-Alderfer, C.; Zheng, Y.; Wang, Q.; Han, X.; Zheng, Q.Y. A new mouse mutant of the Cdh23 gene with early-onset hearing loss facilitates evaluation of otoprotection drugs. Pharmacogenom. J. 2012, 12, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, T.; Lin, F.R.; Someya, S.; Kashio, A.; Sakamoto, T.; Kondo, K. Current concepts in age-related hearing loss: Epidemiology and mechanistic pathways. Hear. Res. 2013, 303, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Schrepfer, T.; Schacht, J. Age-related hearing impairment and the triad of acquired hearing loss. Front. Cell. Neurosci. 2015, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Straatman, L.V.; Lea, J.; Westerberg, B. Current insights in noise-induced hearing loss: A literature review of the underlying mechanism, pathophysiology, asymmetry, and management options. J. Otolaryngol. Head Neck Surg. 2017, 46, 41. [Google Scholar] [CrossRef] [PubMed]
- Kalinec, G.M.; Lomberk, G.; Urrutia, R.A.; Kalinec, F. Resolution of cochlear inflammation: Novel target for preventing or ameliorating drug-, noise- and age-related hearing loss. Front. Cell. Neurosci. 2017, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. Hereditary hearing loss and deafness overview. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mefford, H.C., Stephens, K., Amemiya, A., Ledbetter, N., Eds.; University of Washington: Seattle, WA, USA, 1993–2017. [Google Scholar]

{kind=link}
| Hearing loss | Alteration in ER Stress | Models | ER Stress-Related Treatment | References |
|---|---|---|---|---|
| DIHL | ||||
| 3-NP | ATF4 ↑, CHOP ↑ | Rat/guinea pig cochlea | GGA | [39,56] |
| APAP | p-eIF2α ↑, CHOP ↑ | HEI-OC1 cell | [40] | |
| NAPQI | p-eIF2α ↓, CHOP ↑ | HEI-OC1 cell | [40] | |
| Aminoglycosides | All 3 UPR pathways ↑ | HEK cell, mouse cochlea | TUDCA | [41] |
| NIHL | Bip ↑, CHOP ↑ | Guinea pig/mouse cochlea | SA4503,GGA | [44,45,50,55] |
| ARHL | Bip ↓, CHOP ↑ | Mouse cochlea | GGA | [51,54] |
| Deafness Gene | Alteration in ER Stress | Models | ER Stress-Related Treatment | References |
|---|---|---|---|---|
| WFS1 | ATF6α pathway ↑ | Knockdown cells | [63,64] | |
| All 3 UPR pathways ↑ | Knockout mouse pancreas | [64,71] | ||
| IRE1α pathway ↑ | Knockout mouse retina | [72] | ||
| ATF6α pathway ↑ | Mutant overexpressed cells | [78,79,80] | ||
| GJB3 | All 3 UPR pathways ↑ | Mutant overexpressed cells * | [95] | |
| Bip ↑ | Mutant overexpressed cells | [101] | ||
| CDH23 | PERK pathway ↑ | Mutant mouse cochlea | Salubrinal | [61] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, X.; Xu, Z. Endoplasmic Reticulum Stress in Hearing Loss. J. Otorhinolaryngol. Hear. Balance Med. 2018, 1, 3. https://doi.org/10.3390/ohbm1010003
Wang Y, Liu X, Xu Z. Endoplasmic Reticulum Stress in Hearing Loss. Journal of Otorhinolaryngology, Hearing and Balance Medicine. 2018; 1(1):3. https://doi.org/10.3390/ohbm1010003
Chicago/Turabian StyleWang, Yanfei, Xiangguo Liu, and Zhigang Xu. 2018. "Endoplasmic Reticulum Stress in Hearing Loss" Journal of Otorhinolaryngology, Hearing and Balance Medicine 1, no. 1: 3. https://doi.org/10.3390/ohbm1010003
APA StyleWang, Y., Liu, X., & Xu, Z. (2018). Endoplasmic Reticulum Stress in Hearing Loss. Journal of Otorhinolaryngology, Hearing and Balance Medicine, 1(1), 3. https://doi.org/10.3390/ohbm1010003