SU-QMI: A Feature Selection Method Based on Graph Theory for Prediction of Antimicrobial Resistance in Gram-Negative Bacteria †


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Data Collection
2.2. Protein Features
2.3. Feature Selection
| Algorithm 1: SU-QMI algorithm |
| Input: A complete graph G = (V, E) where V is the set of all features and E denotes the edges representing the normalized interdependency or redundancy value between vertices (features), feature set F, class C, number of features to be selected k, and queue Q. |
| Output: Best feature subset Q |
 |
2.4. Data and Code Availability
3. Results
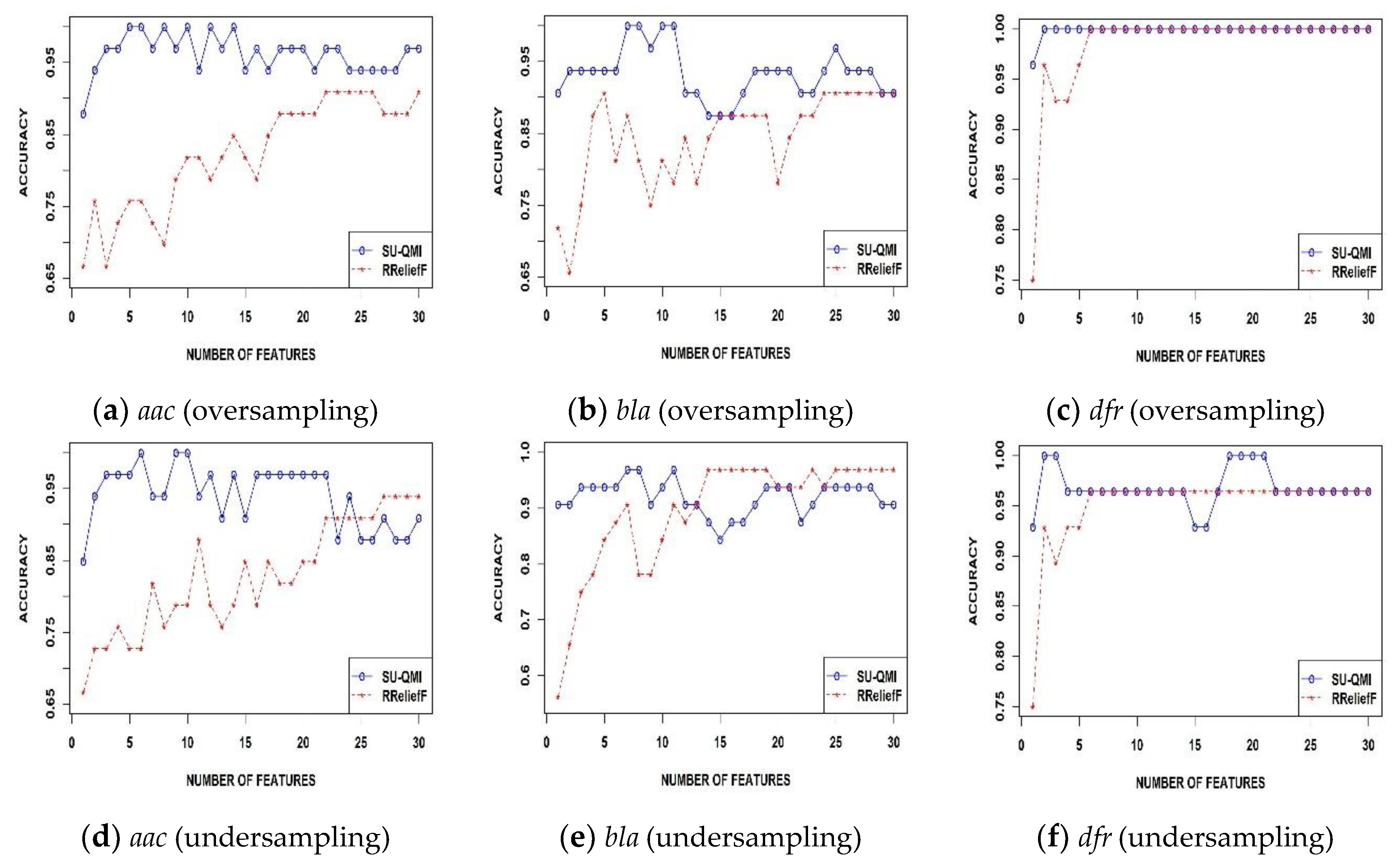
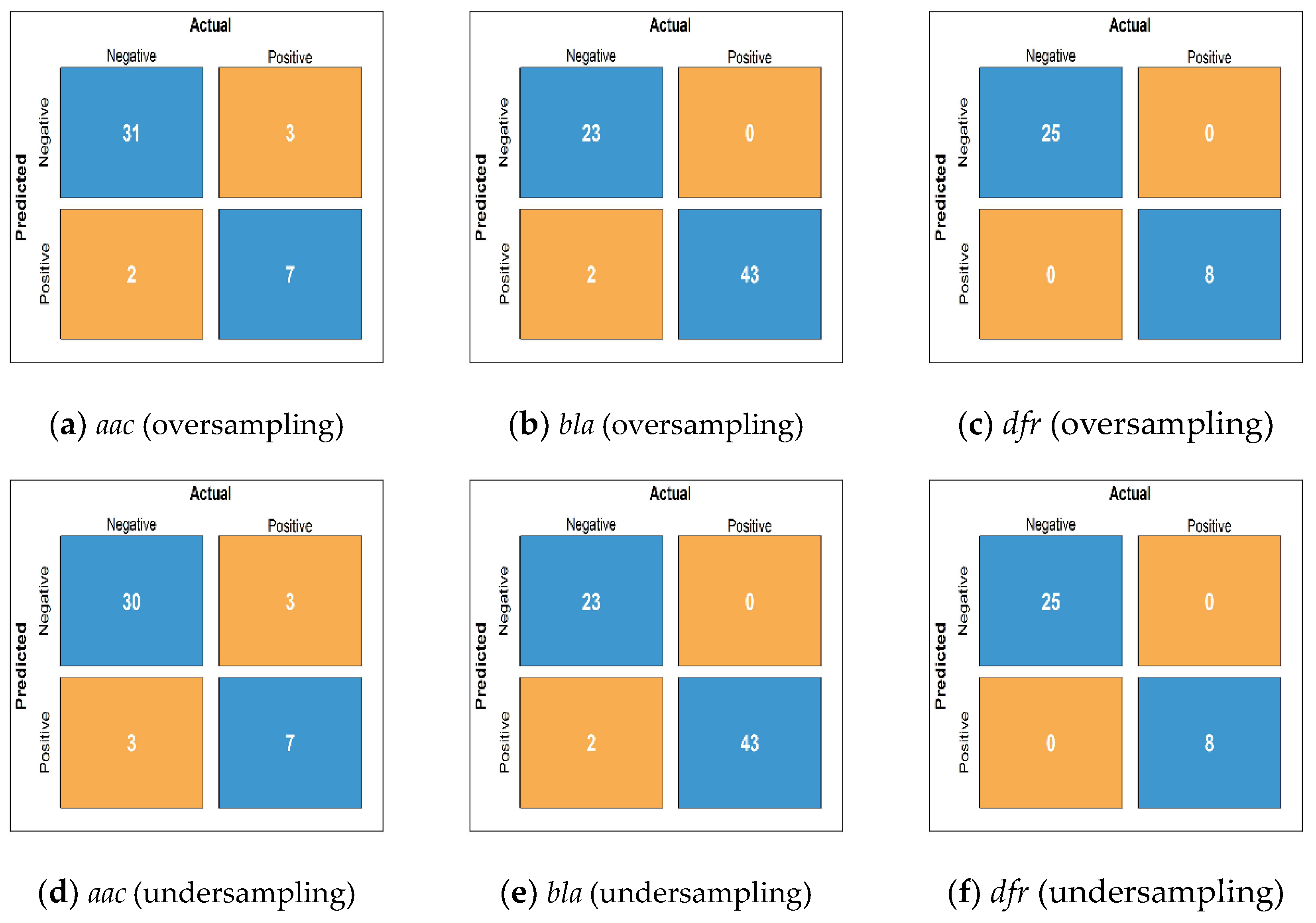
3.1. Comparative Analysis of the SU-QMI Feature Selection Method
3.2. Identification of Antimicrobial-Resistance Proteins in Independent Datasets
4. Discussion
Funding
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). Antibiotic Resistance Threats in the United States 2019; Centers for Disease Control and Prevention, US Department of Health and Human Services: Atlanta, USA, 2019. [Google Scholar]
- Chowdhury, A.S.; Lofgren, E.T.; Moehring, R.W.; Broschat, S.L. Identifying predictors of antimicrobial exposure in hospitalized patients using a machine learning approach. J. Appl. Microbiol. 2020, 128, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Call, D.R.; Broschat, S.L. Antimicrobial Resistance Prediction for Gram-Negative Bacteria via Game Theory-Based Feature Evaluation. Sci. Rep. 2019, 9, 14487. [Google Scholar] [CrossRef] [PubMed]
- Press, W.H.; Teukolsky, S.; Vetterling, W.; Flannery, B.P. Numerical Recipes in C: The Art of Scientific Computing (10.5); Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Luan, H.; Qi, F.; Shen, D. Multi-modal image registration by quantitative-qualitative measure of mutual information (q-mi). In International Workshop on Computer Vision for Biomedical Image Applications; Springer: Berlin/Heidelberg, Germany, 2005; pp. 378–387. [Google Scholar]
- Robnik-Šikonja, M.; Kononenko, I. Theoretical and empirical analysis of ReliefF and RReliefF. Mach. Learn. 2003, 53, 23–69. [Google Scholar] [CrossRef]
- Chowdhury, A.S.; Khaledian, E.; Broschat, S.L. Capreomycin resistance prediction in two species of Mycobacterium using a stacked ensemble method. J. Appl. Microbiol. 2019, 127, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Call, D.R.; Broschat, S.L. PARGT: A software tool for predicting antimicrobial resistance in bacteria. Sci. Rep. 2020, 10, 11033. [Google Scholar] [CrossRef] [PubMed]
- Dubchak, I.; Muchnik, I.; Mayor, C.; Dralyuk, I.; Kim, S.H. Recognition of a protein fold in the context of the SCOP classification. Proteins Struct. Funct. Bioinform. 1999, 35, 401–407. [Google Scholar] [CrossRef]
- Gini, C. Concentration and dependency ratios. Riv. Politica Econ. 1997, 87, 769–792. [Google Scholar]
- Barron, F.H.; Barrett, B.E. Decision quality using ranked attribute weights. Manag. Sci. 1996, 42, 1515–1523. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhury, A.S.; Call, D.R.; Broschat, S.L. SU-QMI: A Feature Selection Method Based on Graph Theory for Prediction of Antimicrobial Resistance in Gram-Negative Bacteria. Proceedings 2020, 66, 7. https://doi.org/10.3390/proceedings2020066007
Chowdhury AS, Call DR, Broschat SL. SU-QMI: A Feature Selection Method Based on Graph Theory for Prediction of Antimicrobial Resistance in Gram-Negative Bacteria. Proceedings. 2020; 66(1):7. https://doi.org/10.3390/proceedings2020066007
Chicago/Turabian StyleChowdhury, Abu Sayed, Douglas R. Call, and Shira L. Broschat. 2020. "SU-QMI: A Feature Selection Method Based on Graph Theory for Prediction of Antimicrobial Resistance in Gram-Negative Bacteria" Proceedings 66, no. 1: 7. https://doi.org/10.3390/proceedings2020066007
APA StyleChowdhury, A. S., Call, D. R., & Broschat, S. L. (2020). SU-QMI: A Feature Selection Method Based on Graph Theory for Prediction of Antimicrobial Resistance in Gram-Negative Bacteria. Proceedings, 66(1), 7. https://doi.org/10.3390/proceedings2020066007