Sequence-Based Discovery of Antibacterial Peptides Using Ensemble Gradient Boosting †


Abstract
:1. Introduction
2. Methods
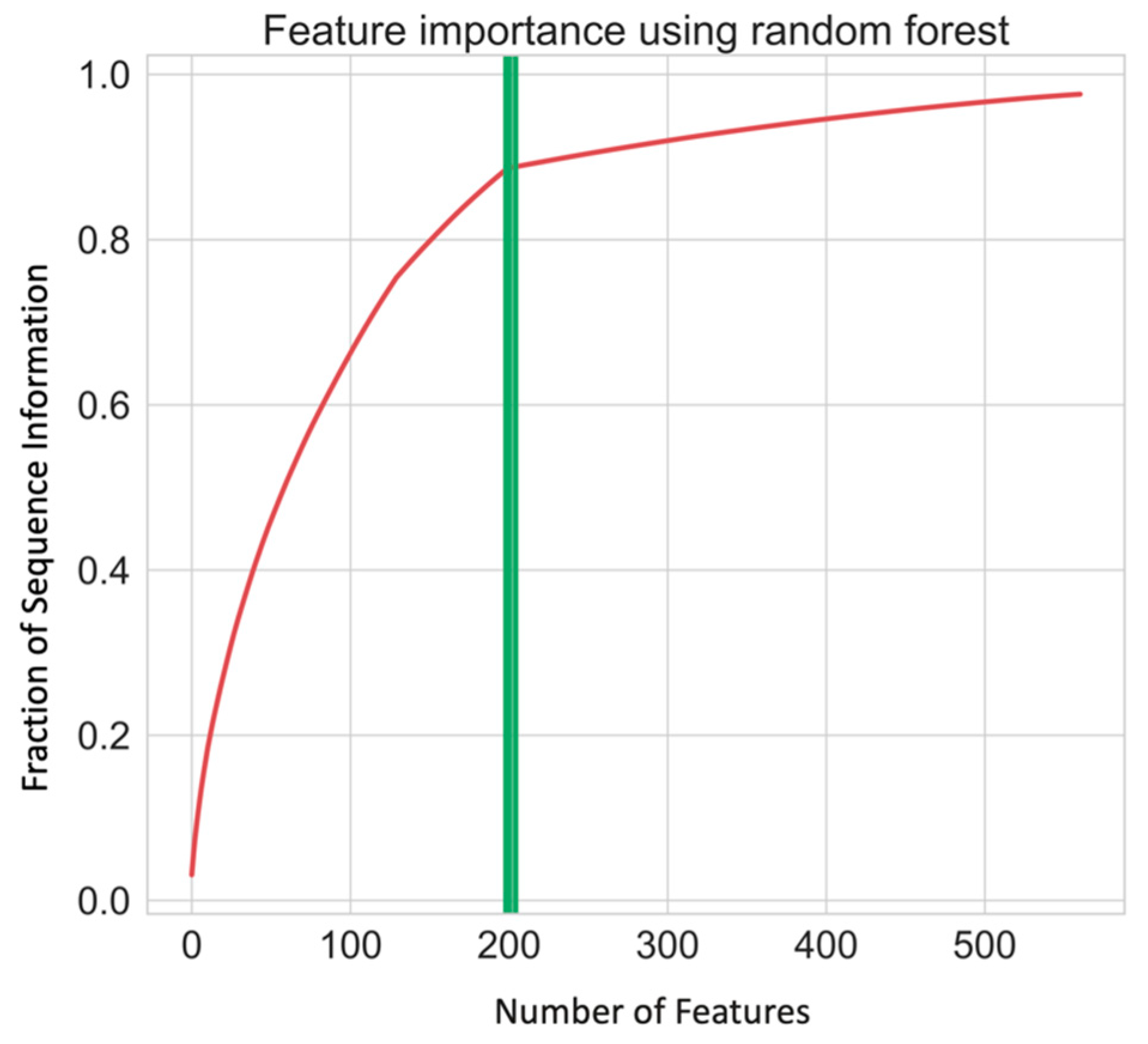
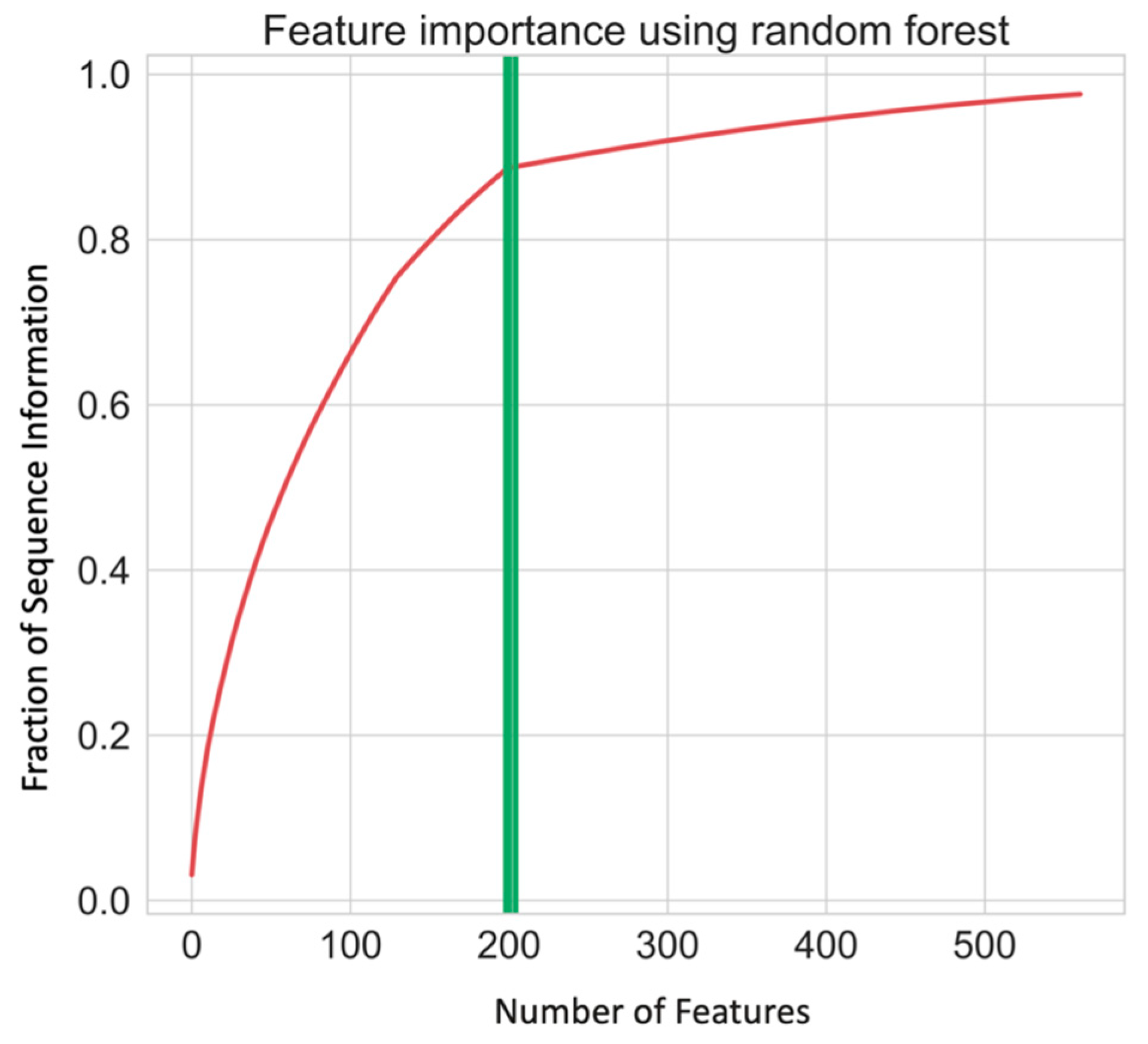
2.1. Data Collection and Feature Selection
2.2. Ensemble Gradient Boosting Model
| Algorithm 1: Gradient Boosting Algorithm |
 |
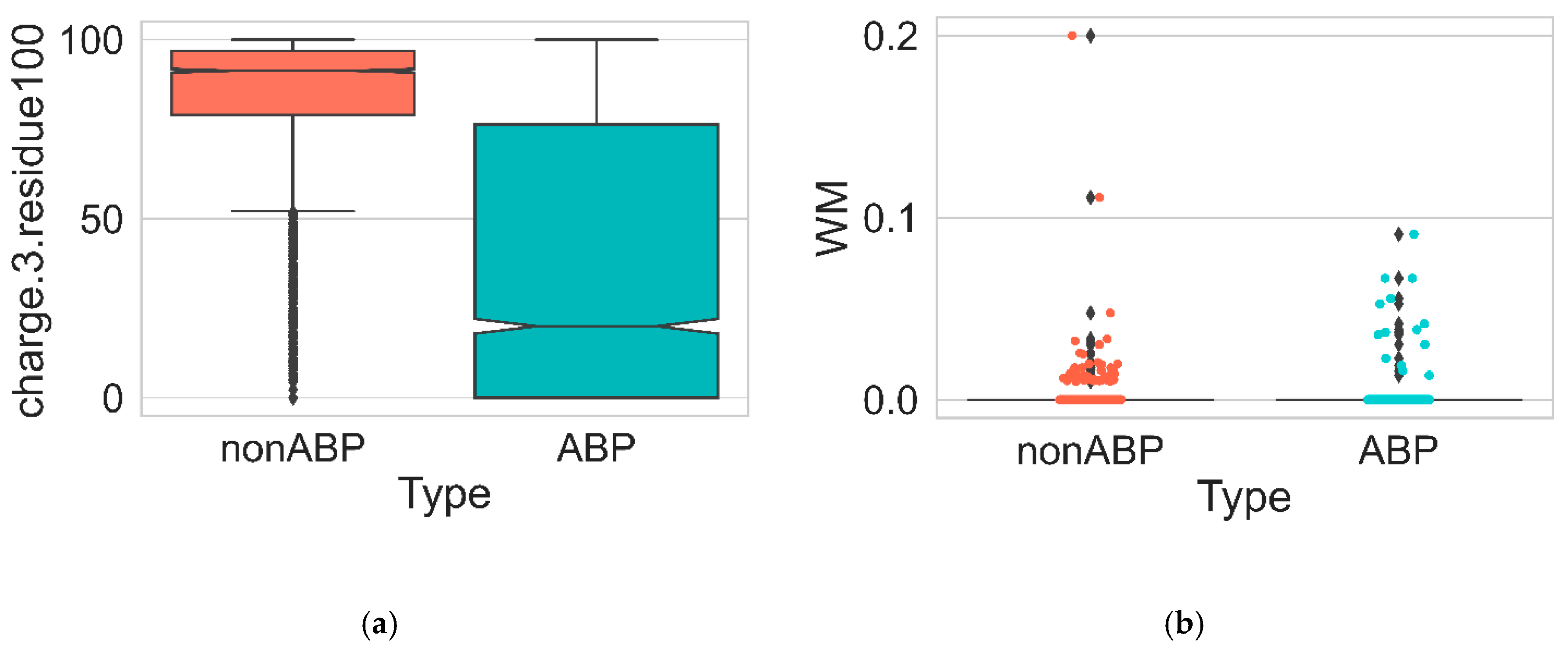
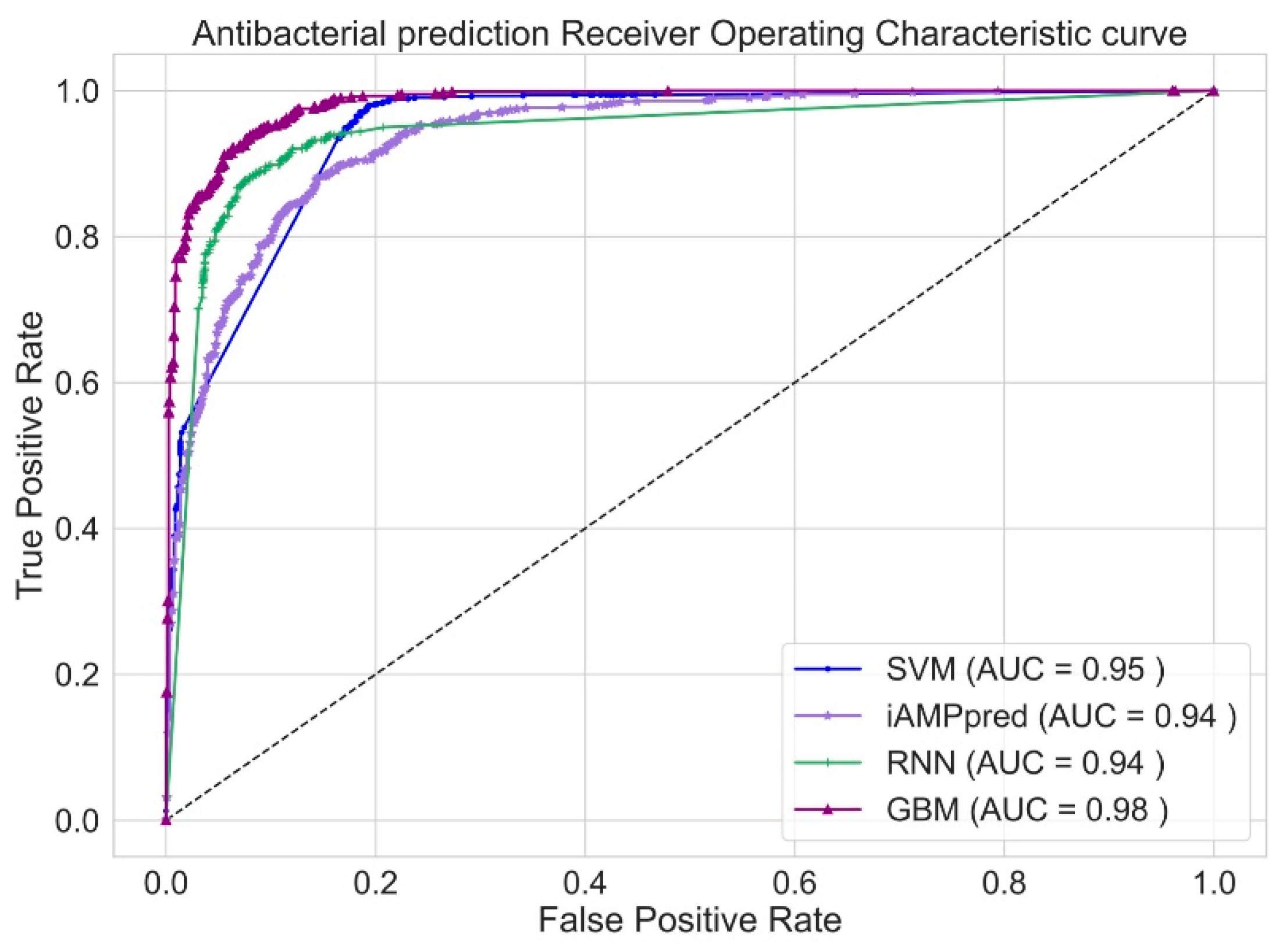
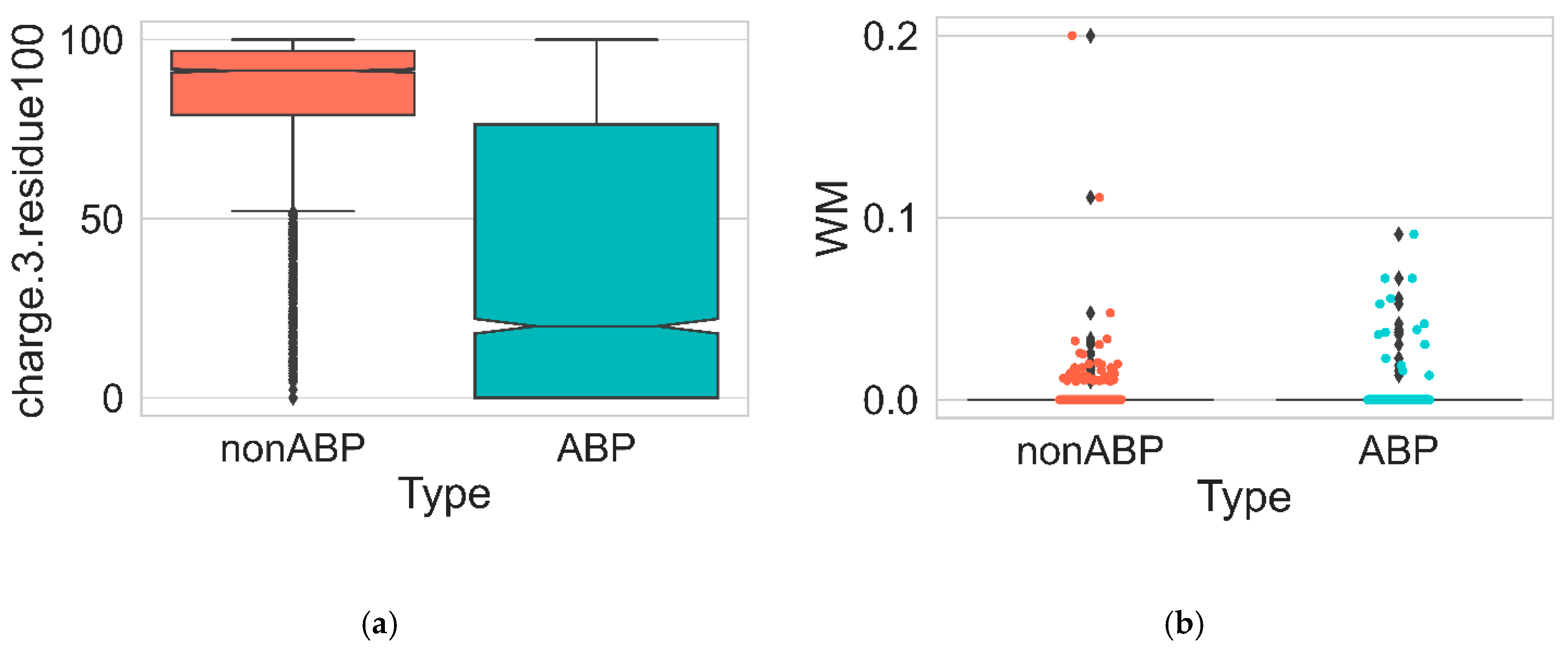
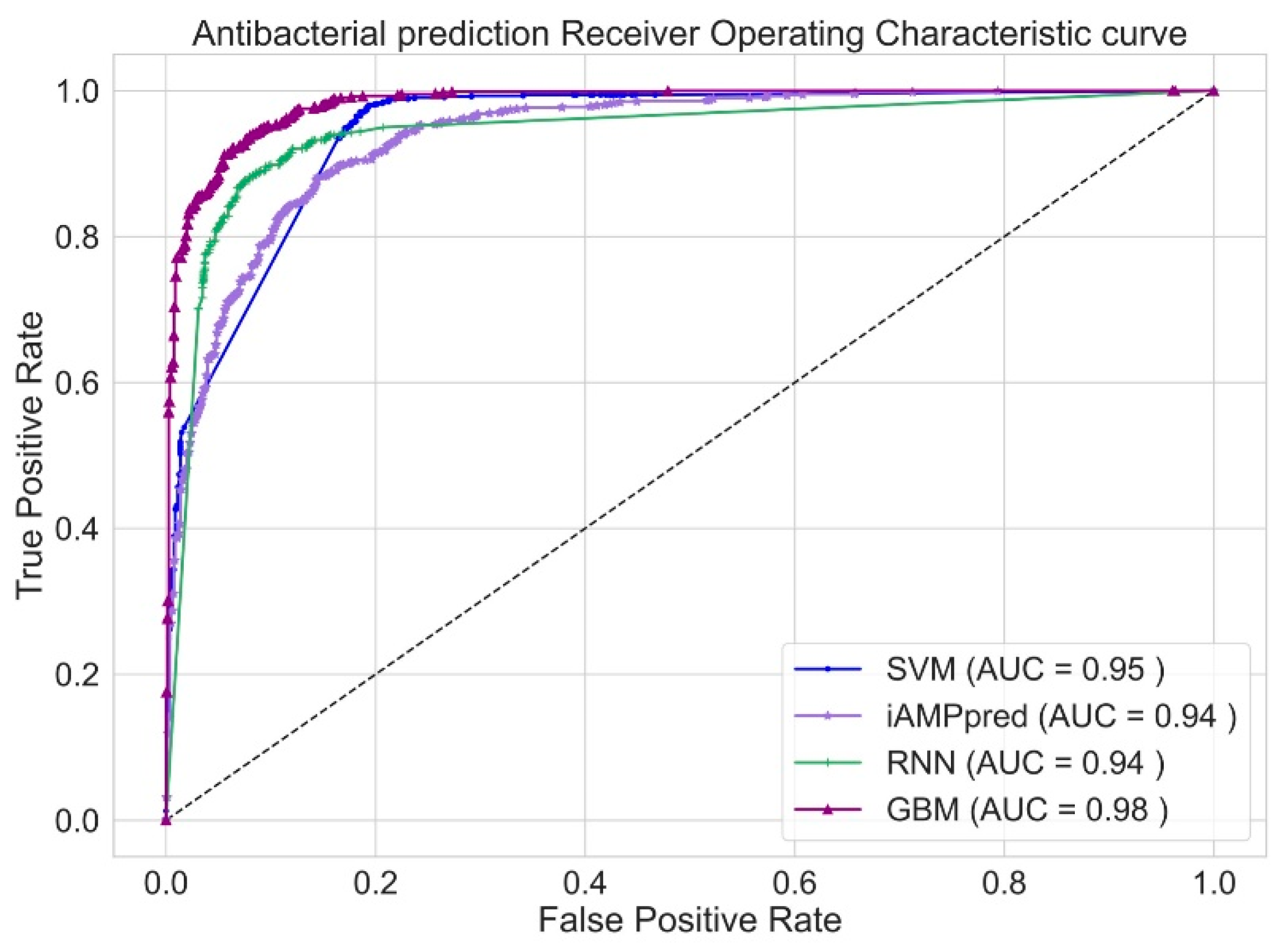
3. Results and Discussion
4. Conclusions
Conflicts of Interest
Abbreviations
| AMP | Antimicrobial peptides |
| ABP | Antibacterial peptides |
| GBM | Gradient Boosting Machine |
References
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Khaledian, E.; Brayton, K.A.; Broschat, S.L. A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science. Microorganisms 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Cao, Z.; Li, M.; Wang, S. ACEP: Improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Patocˇka, J.; Kucˇa, K. Insect antimicrobial peptides, a mini review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Managadze, G.; Grigolava, M.; Makhatadze, G.I.; Pirtskhalava, M. Predictive model of linear antimicrobial peptides active against gram-negative bacteria. J. Chem. Inf. Model. 2018, 58, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. mAHTPred: A sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 2019, 35, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W. Deep-AmPEP30: Improve short antimicrobial peptides prediction with deep learning. Mol. Ther. Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.C.; et al. iFeature: A python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y. Random forest for bioinformatics. In Ensemble Machine Learning; Springer: Berlin/Heidelberg, Germany, 2012; pp. 307–323. [Google Scholar]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- McClish, D.K. Analyzing a portion of the ROC curve. Med. Decis. Mak. 1989, 9, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Feature | Description | Dimension |
|---|---|---|
| AAC | Amino acid composition | 20 |
| DPC | Dipeptide composition | 400 |
| DDE | Dipeptide deviation from expected mean | 400 |
| GAAC | Grouped amino acid composition | 5 |
| GDPC | Grouped dipeptide composition | 25 |
| GTPC | Grouped tripeptide composition | 125 |
| CTDC | Composition | 39 |
| CTDD | Distribution | 195 |
| Total Number of Features | 1209 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaledian, E.; Broschat, S.L. Sequence-Based Discovery of Antibacterial Peptides Using Ensemble Gradient Boosting. Proceedings 2020, 66, 6. https://doi.org/10.3390/proceedings2020066006
Khaledian E, Broschat SL. Sequence-Based Discovery of Antibacterial Peptides Using Ensemble Gradient Boosting. Proceedings. 2020; 66(1):6. https://doi.org/10.3390/proceedings2020066006
Chicago/Turabian StyleKhaledian, Ehdieh, and Shira L. Broschat. 2020. "Sequence-Based Discovery of Antibacterial Peptides Using Ensemble Gradient Boosting" Proceedings 66, no. 1: 6. https://doi.org/10.3390/proceedings2020066006
APA StyleKhaledian, E., & Broschat, S. L. (2020). Sequence-Based Discovery of Antibacterial Peptides Using Ensemble Gradient Boosting. Proceedings, 66(1), 6. https://doi.org/10.3390/proceedings2020066006