Attainment of Pentagonal-Bipyramidal LnIII Complexes from a Planar Pentadentate Ligand †





Abstract
:1. Introduction
2. Materials and Methods
2.1. Syntheses of the Complexes
2.2. X-ray Studies
2.3. Magnetic Measurements
3. Results and Discussion
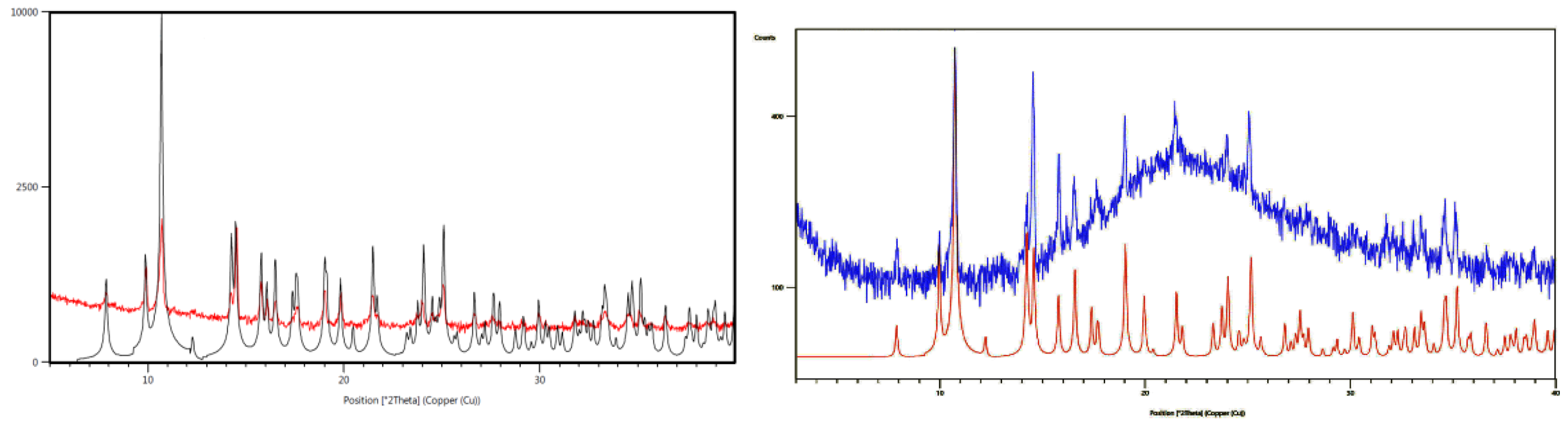
3.1. Crystal Structures
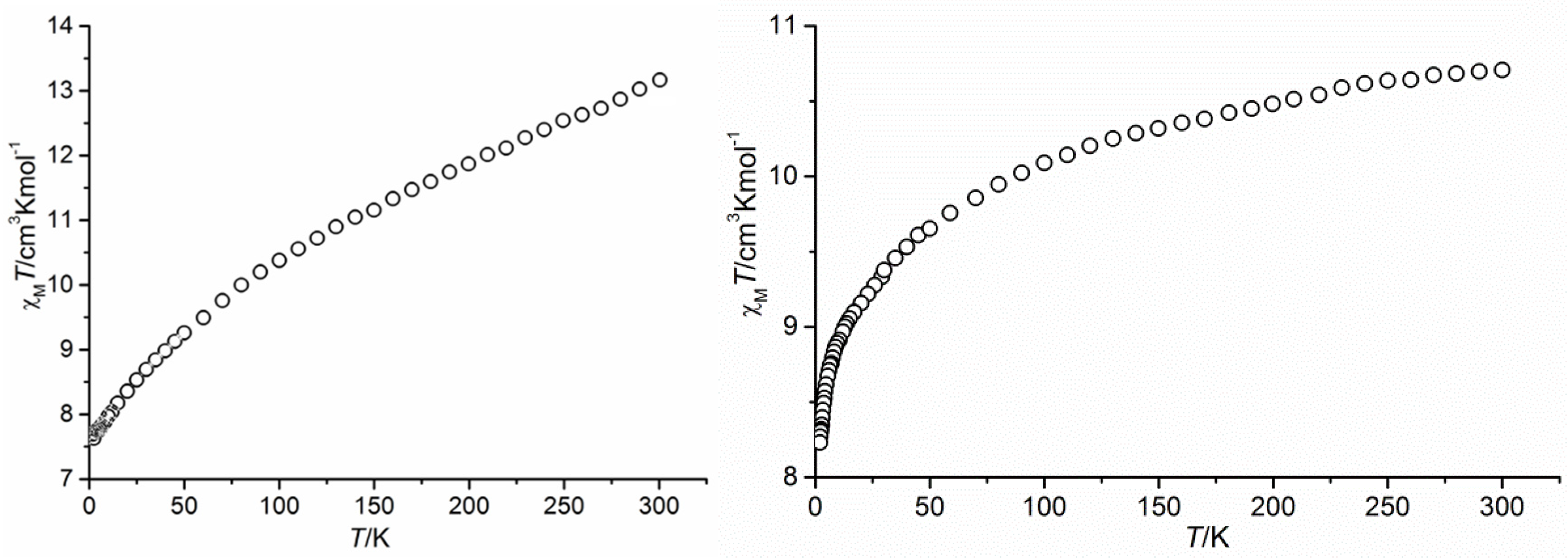
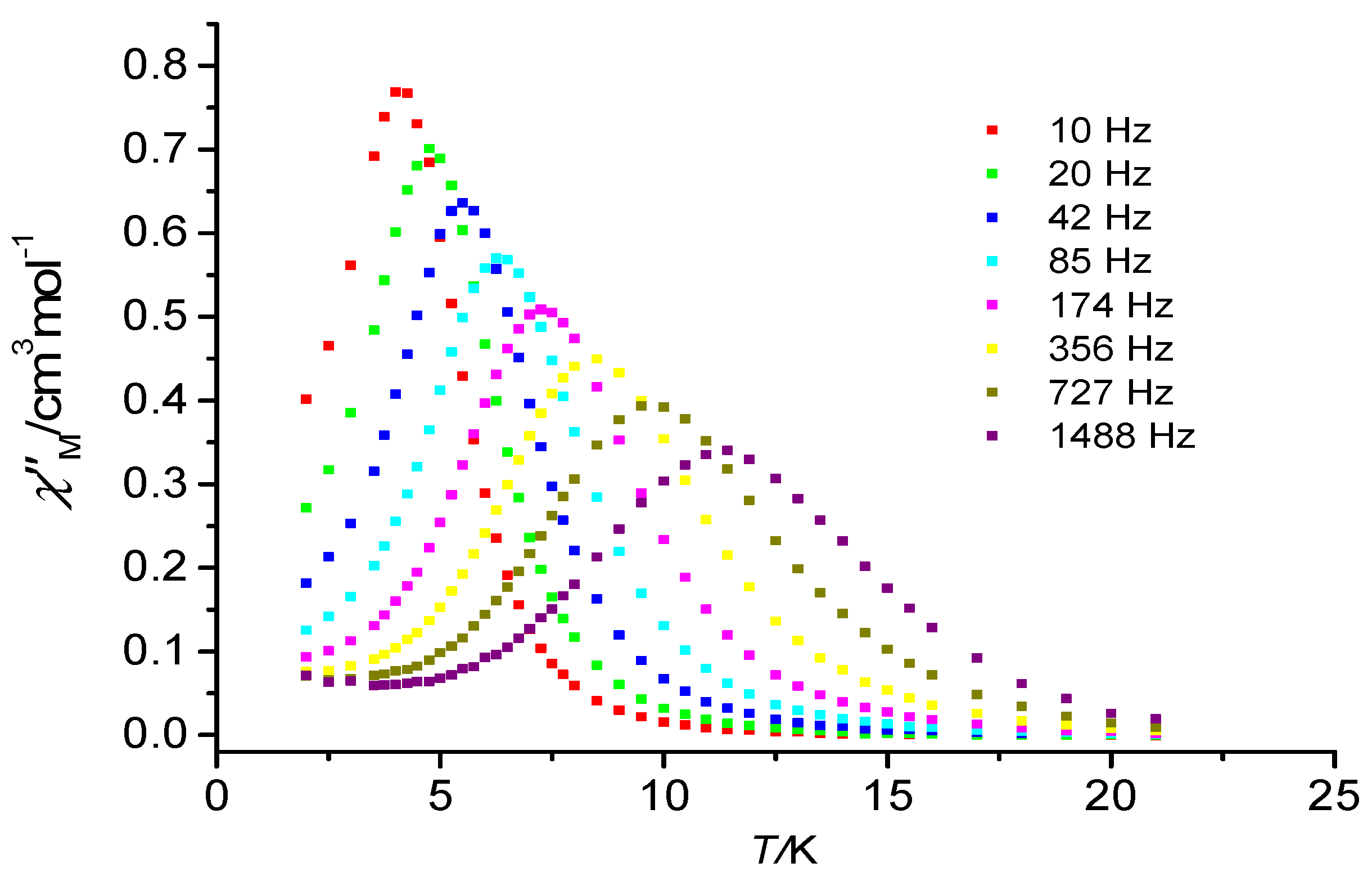
3.2. Magnetic Properties
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Lanthanide double decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2011, 2, 2078–2085. [Google Scholar] [CrossRef]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Liu, J.-L.; Ungur, L.; Liu, J.; Li, Q.-W.; Wang, L.-F.; Ni, Z.-P.; Chibotaru, L.F.; Chen, X.-M.; Tong, M.-L. Symmetry-supported magnetic blocking at 20 K in pentagonal bipyramidal Dy(III) single-ion magnets. J. Am. Chem. Soc. 2016, 138, 2829–2837. [Google Scholar] [CrossRef] [PubMed]
- Pelizzi, C.; Pelizzi, G. Investigation into aroylhydrazones as chelating agents. Synthesis and structural characterization of a tin(IV) complex with 2,6 diacetylpyridine bis(salicyloylhydrazone). J. Chem. Soc. Dalton Trans. 1980, 1970–1973. [Google Scholar] [CrossRef]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, A51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Bar, A.K.; Kalita, P.; Sutter, J.-P.; Chandrasekhar, V. Pentagonal-bipyramid Ln(III) complexes exhibiting single-ion-magnet behavior: A rational synthetic approach for a rigid equatorial plane. Inorg. Chem. 2018, 57, 2398–2401. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, A.; Merlo, C.; Pelizzi, C.; Pelizzi, G.; Tarasconi, P.; Cavatorta, F. Synthesis, spectroscopic and structural characterization of mono- and binuclear iron(II) complexes with 2,6-diacetylpyridine bis(acylhydrazones). J. Chem. Soc. Dalton Trans. 1991, 1063–1069. [Google Scholar] [CrossRef]
- Bermejo, M.R.; Fondo, M.; Gonzalez, A.M.; Hoyos, O.L.; Sousa, A.; McAuliffe, C.A.; Hussain, W.; Pritchard, R.; Novotorsev, V.M. Electrochemical synthesis and structural characterization of transition metal complexes with 2,6-bis(1-salicyloylhydrazonoethyl)pyridine, H4daps. J. Chem. Soc. Dalton Trans. 1999, 2211–2218. [Google Scholar] [CrossRef]
- Fondo, M.; Corredoira-Vázquez, J.; García-Deibe, A.M.; Sanmartín-Matalobos, J.; Herrera, J.M.; Colacio, E. Tb2, Dy2, and Zn2Dy4 Complexes Showing the Unusual Versatility of a Hydrazone Ligand toward Lanthanoid Ions: A Structural and Magnetic Study. Inorg. Chem. 2018, 57, 10100–10110. [Google Scholar] [CrossRef] [PubMed]
- Bolotina, N.B. X-ray diffraction analysis of modulated crystals: Review. Crystallogr. Rep. 2007, 52, 647–658. [Google Scholar] [CrossRef]
- Mondal, A.K.; Goswami, S.; Konar, S. Influence of the coordination environment on slow magnetic relaxation and photoluminescence behavior in two mononuclear dysprosium(III) based single molecule magnets. Dalton Trans. 2015, 44, 5086–5094. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (CH3)4N[Dy(H2daps)Cl2] | (CH3)4N[Er(H2daps)Cl2] | |
|---|---|---|
| Formula | C27H31Cl2DyN6O4 | C27H31Cl2ErN6O4 |
| Molecular Weight | 736.98 | 741.74 |
| Temperature (K) | 225(2) | 250(2) |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | C m c 21 | C m c 21 |
| a (Å) | 17.8879(9) | 17.8142(18) |
| b (Å) | 14.3964(7) | 14.5177(14) |
| c (Å) | 12.2189(6) | 12.1790(14) |
| Volume (Å3) | 3146.6(3) | 3149.7(6) |
| Z | 4 | 4 |
| Absorp. Coef. (mm−1) | 2.585 | 2.874 |
| Reflections collected | 28,494 | 17,722 |
| Independent reflections | 3529 (Rint = 0.0353) | 4710 (Rint = 0.0391) |
| Data/restraints/param. | 3529/1/200 | 4710/1/201 |
| Final R indices [I > 2σ(I)] | R1 = 0.0268, wR2 = 0.0534 | R1 = 0.0327, wR2 = 0.0654 |
| R indices (all data) | R1 = 0.0328, wR2 = 0.0560 | R1 = 0.0420, wR2 = 0.0691 |
| Dy1-O11 | 2.253(4) | Er1-O11 | 2.238(4) |
|---|---|---|---|
| Dy1-N12 | 2.426(8) | Er1-N12 | 2.418(8) |
| Dy1-N11 | 2.449(5) | Er1-N13 | 2.426(5) |
| Dy1-Cl2 | 2.613(2) | Er1-Cl1 | 2.585(3) |
| Dy1-Cl1 | 2.623(2) | Er1-Cl2 | 2.604(3) |
| Cl2-Dy1-Cl1 | 171.44(9) | Cl2-Er1-Cl1 | 171.73(10) |
| O11#1-Dy1-O11 | 98.94(18) | O11#1-Er1-O11 | 96.36(19) |
| O11-Dy1-N11 | 65.38(15) | O11-Er1-N11 | 66.00(15) |
| N12-Dy1-N11 | 65.17(11) | N11-Er1-N12 | 65.85(12) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corredoira-Vázquez, J.; Fondo, M.; Sanmartín-Matalobos, J.; García-Deibe, A.M. Attainment of Pentagonal-Bipyramidal LnIII Complexes from a Planar Pentadentate Ligand. Proceedings 2020, 62, 2. https://doi.org/10.3390/proceedings2020062002
Corredoira-Vázquez J, Fondo M, Sanmartín-Matalobos J, García-Deibe AM. Attainment of Pentagonal-Bipyramidal LnIII Complexes from a Planar Pentadentate Ligand. Proceedings. 2020; 62(1):2. https://doi.org/10.3390/proceedings2020062002
Chicago/Turabian StyleCorredoira-Vázquez, Julio, Matilde Fondo, Jesús Sanmartín-Matalobos, and Ana M. García-Deibe. 2020. "Attainment of Pentagonal-Bipyramidal LnIII Complexes from a Planar Pentadentate Ligand" Proceedings 62, no. 1: 2. https://doi.org/10.3390/proceedings2020062002
APA StyleCorredoira-Vázquez, J., Fondo, M., Sanmartín-Matalobos, J., & García-Deibe, A. M. (2020). Attainment of Pentagonal-Bipyramidal LnIII Complexes from a Planar Pentadentate Ligand. Proceedings, 62(1), 2. https://doi.org/10.3390/proceedings2020062002