1. Introduction
Antimicrobial substances can be used to treat animals for preventive or curative purposes. The treatments could lead to the presence of residues because of the distribution of the active substance in the animal body, its metabolization, and its excretion. Therefore, antimicrobial residues could be found in animal derived food products (e.g., milk, meat, eggs, honey, etc.). For authorized antimicrobials, maximum residue limits (MRL) have been established for consumer protection based on toxicological data and food consumption [
1]. Nevertheless, some antimicrobials with proven toxicity have been banned for use in livestock animals. For banned substances, minimum required performance limits (MRPLs) have been defined [
2]. MRPLs correspond to the minimum content of an analyte in a sample, which at least has to be detected and confirmed. In order to monitor the presence of these residues in different foodstuffs, surveillance and control plans (SCPs) are put in place in each Member State according to Directive 96/23/EC [
3], now replaced by Regulation (EU) No. 2017/625 [
4]. Two types of methods are implemented in monitoring plans: firstly, screening methods, which generally give a qualitative result (positive (suspect) or negative (compliant)). If the result is positive, a confirmation step is required to identify and quantify the antimicrobial residues. Confirmatory methods are physico-chemical methods based on liquid chromatography coupled mass spectrometry in tandem (LC-MS/MS). MRPLs have been set to harmonize the analytical performance of these methods.
The screening step is crucial because it is the first step of the control. Screening methods should be cheap, quick, sensitive (<5% of false negative results), specific for banned antimicrobials, and with high throughput of samples. Chloramphenicol (CAP) is an efficient, cheap, and wide spectrum antimicrobial. It has been banned since 1993 because of the risks of aplastic anemia and bone marrow suppression [
5,
6]. Bees, like all animals, are susceptible to bacteria, viruses, and parasites (e.g., American foulbrood). Very few treatments are authorized in apiculture. Even authorized antimicrobials for other food-producing animals are not authorized in bee products. Chloramphenicol is also banned in honey and other bee products. However, the illegal use of CAP could lead to the presence of residues in honey. It is of the utmost importance to look for antimicrobial residues in honey. The monitoring of antimicrobials in honey is performed by official control laboratories and by industries for autocontrol purposes. The MRPL of CAP was set at 0.3 µg/kg for all animal-derived food products.
Immunological methods (i.e., ELISA kits, radioimmunoassays (RIA)) and LC-MS/MS methods are commonly used monitoring methods. ELISA tests are available for the screening of CAP in food products at or below the MRPL [
7], and especially in honey [
8,
9,
10]. Historically, immunological methods were the only ones used for the screening of CAP. For twenty years, LC-MS/MS methods have been used more and more for official controls. They have gradually replaced biological ones because they allow for the identification and quantification of a large number of antimicrobials with a single method (broad spectrum), with very good specificity and lower detection levels than those of biological methods. Each ELISA test is able to detect only one single compound [
8,
11]. On the other hand, multi-residue LC-MS/MS methods have been developed for the screening (identification) of several banned substances simultaneously [
12,
13,
14]. However, LC-MS/MS methods need high cost equipment and specifically trained people. These systems are not fitted for field controls. The multiplexing potential of biosensors could be an alternative to LC-MS/MS methods for the simultaneous detection of several antimicrobial residues. A commercial system, named Evidence Investigator™ (Randox, United Kingdom), is based on biochip array technology (BAT) and on chemiluminescence detection. The detection of CAP in honey could be performed using a kit named AM III
® (AntiMicrobial array), which is specific for the detection of CAP or AM V for the simultaneous detection of CAP and nitroimidazoles. However, it is a quite expensive method regarding the investment in the system and the cost of the kits. On the contrary, our project aimed to develop a cheap and sensitive biosensor for the detection of CAP. The perspective will be to develop an innovative multiplex amperometric biosensor for the simultaneous detection of CAP and nitrofuran metabolites in honey. Attempts have been made to develop optical biosensors (e.g., surface plasmon resonance (SPR) for the screening of CAP in different food matrices [
15,
16])). However, the high cost of investment has considerably slowed down the field applications of this system. Electrochemical biosensors represent a promising alternative to optical biosensors, especially for food safety applications, because they are cost-effective, quick and portable systems [
17,
18,
19].
Most of the biosensors developed for the detection of CAP were optical (colorimetric, fluorometric) and electrochemical biosensors [
20]. Colorimetric sensors are interesting because of their naked-eye potential, when fluorometric sensors require an instrumental reading. Optical biosensors are sensitive, specific, simple and quick biosensors for CAP detection. The detection capabilities of electrochemical sensors were shown to be lower than those of optical sensors. In particular, voltametric and electrochemiluminescent sensors were the most sensitive. Therefore, we decided to focus this work on electrochemical biosensors as a cheap, sensitive, and selective alternative to ELISA tests and LC-MS/MS methods. Electrochemical biosensors for the detection of antimicrobial residues were firstly developed based on antibodies as recognition elements (i.e., immunosensors). More recently, aptasensors have become more and more prevalent in the literature [
20]. However, very few aptamers against antimicrobials are commercially available. The detection methods for electrochemical biosensors are diverse (i.e., amperometry, potentiometry, field-effect transistors (FETs), conductometry [
21]). A Spanish team worked on the detection of different families of antimicrobial residues in milk by amperometric detection [
22,
23,
24] and finally succeeded to develop a multiplex immunosensor for the detection of three families of antimicrobials simultaneously [
25]. They obtained very promising results regarding the sensitivity of the developed methods. This immunosensor is based on the binding of antibodies against the antimicrobial residue to magnetic beads. Then, the analyte (i.e., the antimicrobial residue) competes with the horseradish peroxidase (HRP)-conjugate (HRP bound to the analyte) to bind to the antibodies. At the end, the electrochemical activity of the enzyme HRP is measured by amperometry on screen-printed carbon electrodes (SPCE). The concentration of antimicrobial residues is inversely proportional to the electrochemical signal because it is a competitive immunoassay. The aim of this work was to develop a similar bead-based amperometric immunosensor for the detection of chloramphenicol in honey.
The aim of this work was to develop and to optimize an amperometric biosensor for the detection of CAP in honey. The project consisted of two steps: optimization of the sensor and then development of an extraction protocol for honey samples to remove matrix interferences. The first results in buffer with simple amperometric conditions were very promising and satisfactory, as well as preliminary results in one single acacia honey sample. Different extraction protocols (liquid/liquid extraction (LLE)) were tested to obtain an optimal detection capability, at or below 0.3 ng/g (CAP MRPL). When analyzing honey samples from different botanical origins, with different colors and textures, high matrix effects were observed. Regarding each individual sample, it was possible to discriminate blank samples from samples spiked with CAP to 0.3 ng/g. However, a global analysis was impossible due to the high variability of honey samples and to high matrix interferences. We started to explore some paths to solve this issue, but a lot of work remains.
2. Materials and Methods
2.1. Antibiotic Standards
Chloramphenicol (CRM) (CAS (Chemical Abstracts Service) number: 56-75-7) was purchased from Sigma Aldrich (Saint-Quentin Fallavier, France).
2.2. Reagents
All buffer solutions were prepared with Milli-Q® water. PBS (Phosphate Buffered Saline) at pH 7.5 contained 0.01 M of phosphate buffer, 137 mM of NaCl (Acros Organics, reference 207790010) and 2.7 mM of KCl (Merck Chemicals, 104936). PBS-T (PBS-Tween) was derived from this PBS solution, to which 0.05% Tween 20 (Sigma Aldrich, Saint-Quentin Fallavier, France, reference P1379) was added. The phosphate buffered solutions at 0.05 M pH 6.0 and 0.1 M pH 6.0 were prepared from Na2HPO4·2H2O (Labosi, Fisher Scientific A4894351) and NaH2PO4 (Merck, 1.06345.1000).
Hydrogen peroxide (H2O2) (Sigma Aldrich, France, reference H-0904) and hydroquinone (Sigma Aldrich, France, reference H9003) were prepared in phosphate buffer.
The antibody anti-chloramphenicol (reference PAS 9681) and the conjugate CAP-HRP (reference HRP9263) were purchased from Randox Life Sciences (UK).
Magnetic beads (MBs) Protein G InVitrogen Dynabeads® (reference 10003D) were purchased from ThermoFisher Scientific (France). The magnetic beads contain protein G on their surface that facilitate the immobilization of antibodies by their Fc regions.
Poly(vinylpolypyrrolidone) (PVPP) was purchased from Sigma Aldrich (France, reference 77627).
2.3. Preparation of Spiked Honey
Honeys from different floral origins and from different suppliers have been collected. These honey samples have already been tested for the presence of antimicrobial residues with AM I and AM II kits (Randox, Crumlin, UK) with the Evidence Investigator™ system.
A stock solution of CAP at 1 mg/mL was prepared in methanol and stored at −20 °C for 1 year. Then, the stock solution was diluted to prepare spiking solutions at different concentrations. Spiking working solutions were prepared day-to-day. The final dilution in honey was always performed by adding 50 µL of spiking solution to 1 g of honey.
2.4. Materials
The potentiostat (STAT300) supplied by Methrom (Herisau, Switzerland) was connected via USB (Universal Serial Bus) to a computer installed with Dropview 8400 software (Methrom, Herisau, Switzerland).
Magnetic rack magnetic support (reference MAGNET16TUBE05) was purchased from DropSens (Oviedo, Spain). The thermoshaker (reference TS-100) was commercialized by BioSan (Guibeville, France).
Non-modified screen-printed carbon electrodes (SPCE) (reference DRP-110) and SPCE modified with carboxyl-functionalized multi-walled carbon nanotubes (MWCNT-COOH) (110CNT) were purchased from Methrom, Switzerland. An SPCE consists of a ceramic substrate incorporating the electrochemical cell. DRP-110 electrodes consisted of working (4 mm diameter) and counter electrodes made of carbon; the reference electrode was silver. The 110CNT carbon nanotubes consisted of a working electrode (4 mm diameter) made of multi-walled carbon nanotubes (MWCNT); the counter electrode was made of carbon, whereas the reference electrode and electric contacts were made of silver.
2.5. Sample Preparation for Analyses
Different extraction procedures for honey samples have been tested to find the optimized procedure. These different procedures are presented with the results.
2.6. Assay Protocol
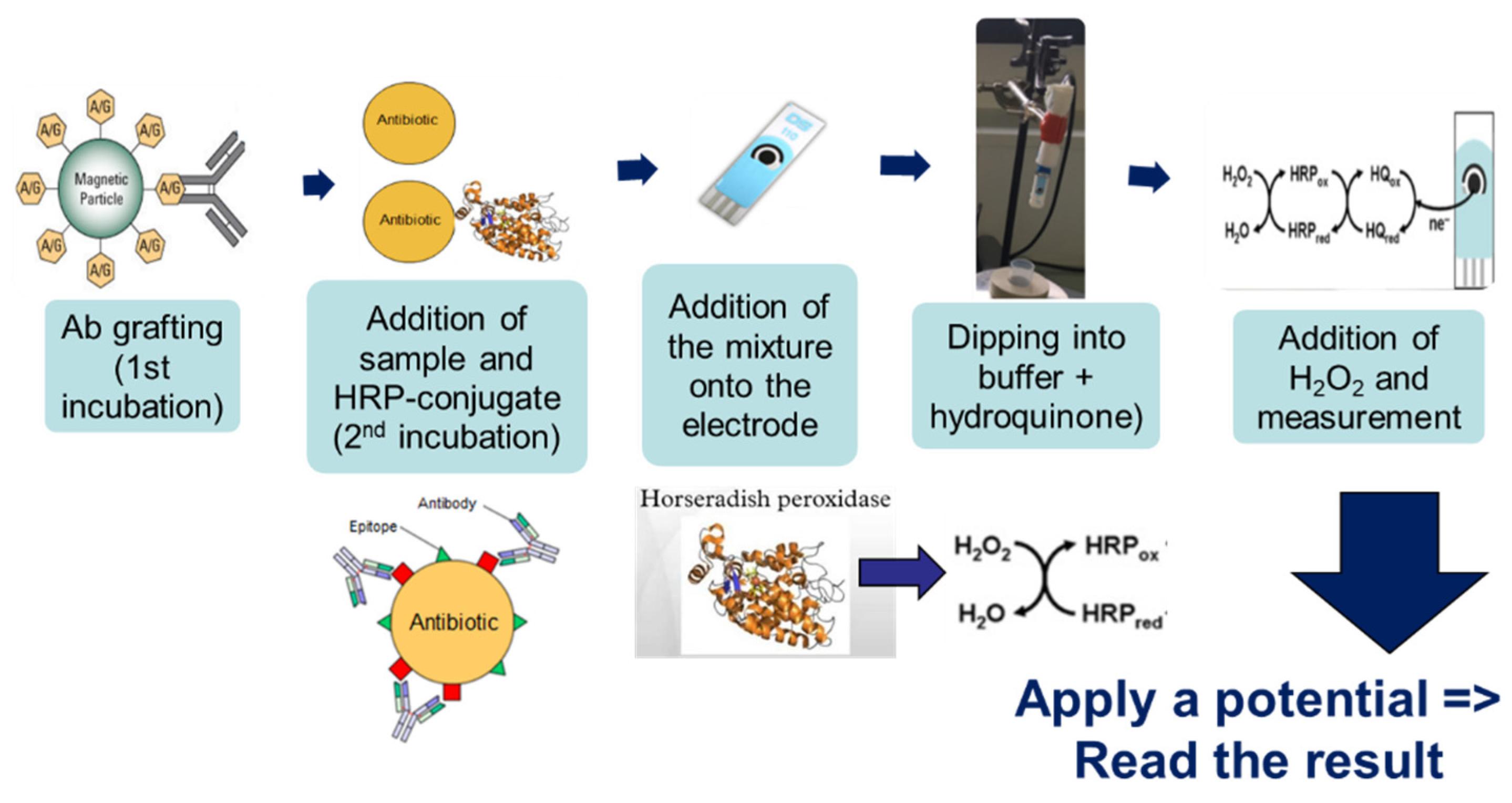
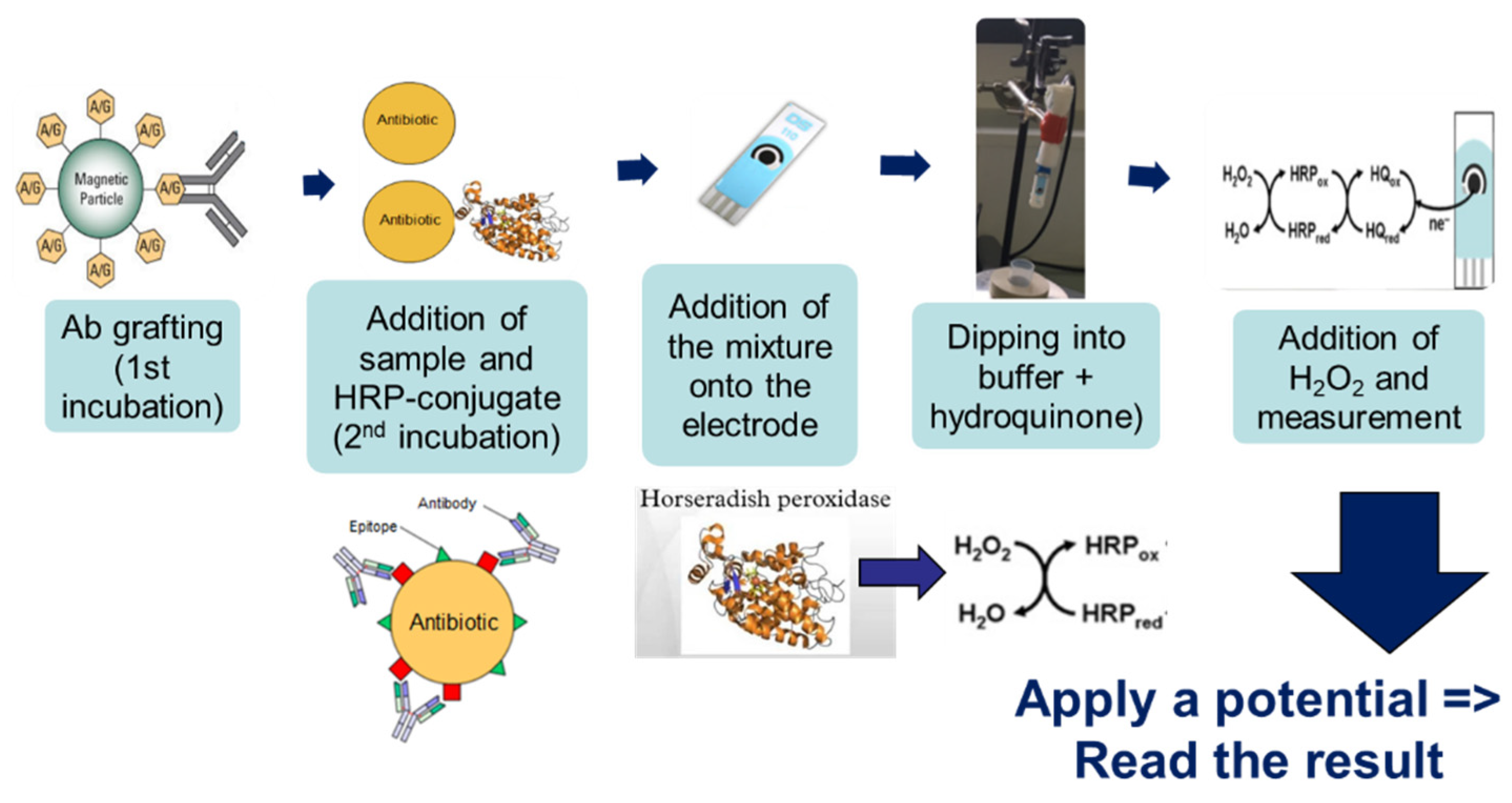
The method’s general principle was based on immuno-competition between the CAP–HRP conjugate and the antimicrobial present (or not) in the sample for binding to the antibodies (Ab) pre-grafted onto magnetic beads (
Figure 1). If there was no CAP in the sample (blank sample), the conjugate was able to bind to the antibody, and so a large quantity of HRP could react with H
2O
2 during the electrochemical reaction, thus generating a large amperometric signal. On the other hand, if CAP was present in the sample, there was competition between CAP and the HRP-conjugate; therefore, less CAP–HRP was bound to Ab–MBs, and the amperometric signal decreased.
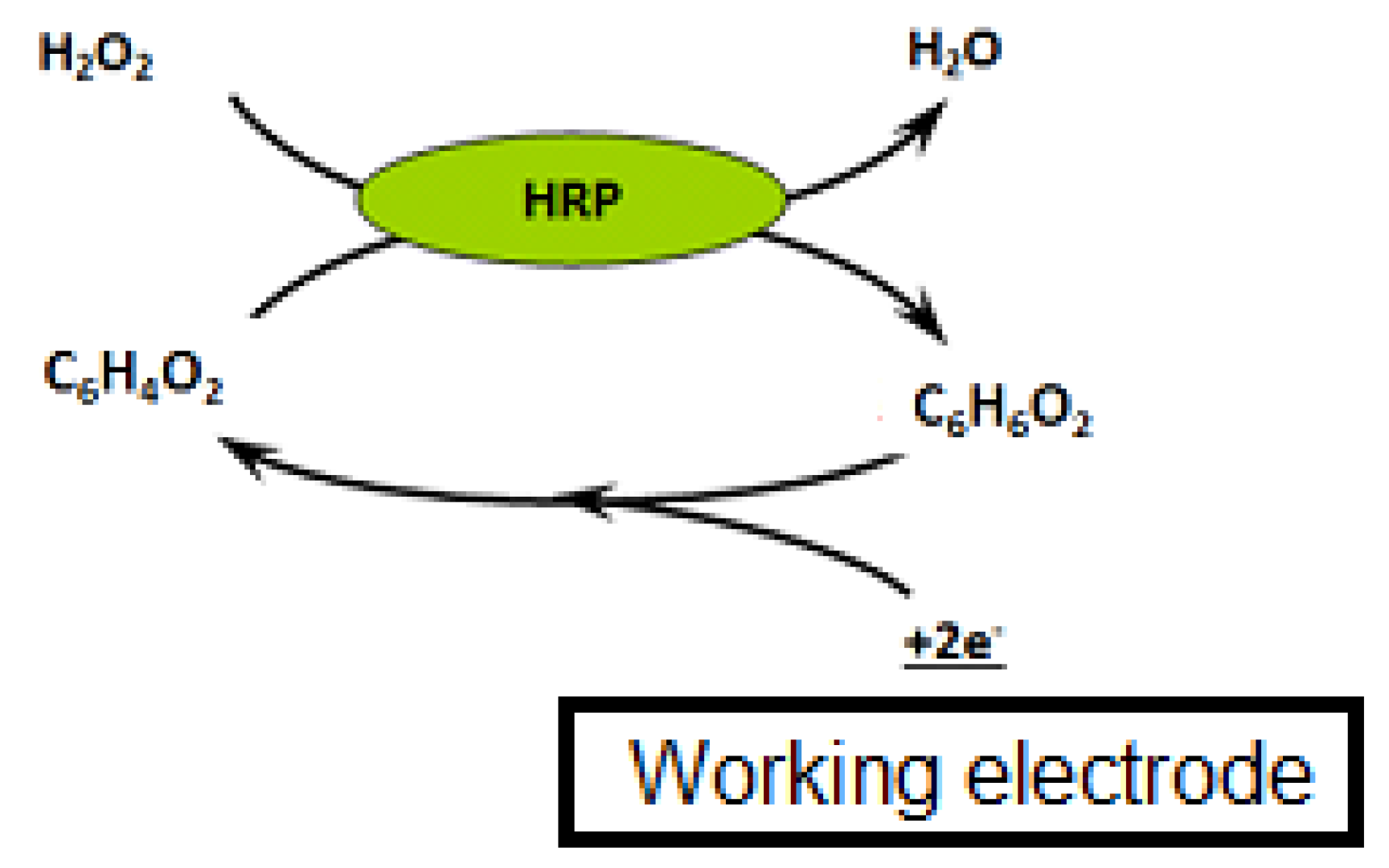
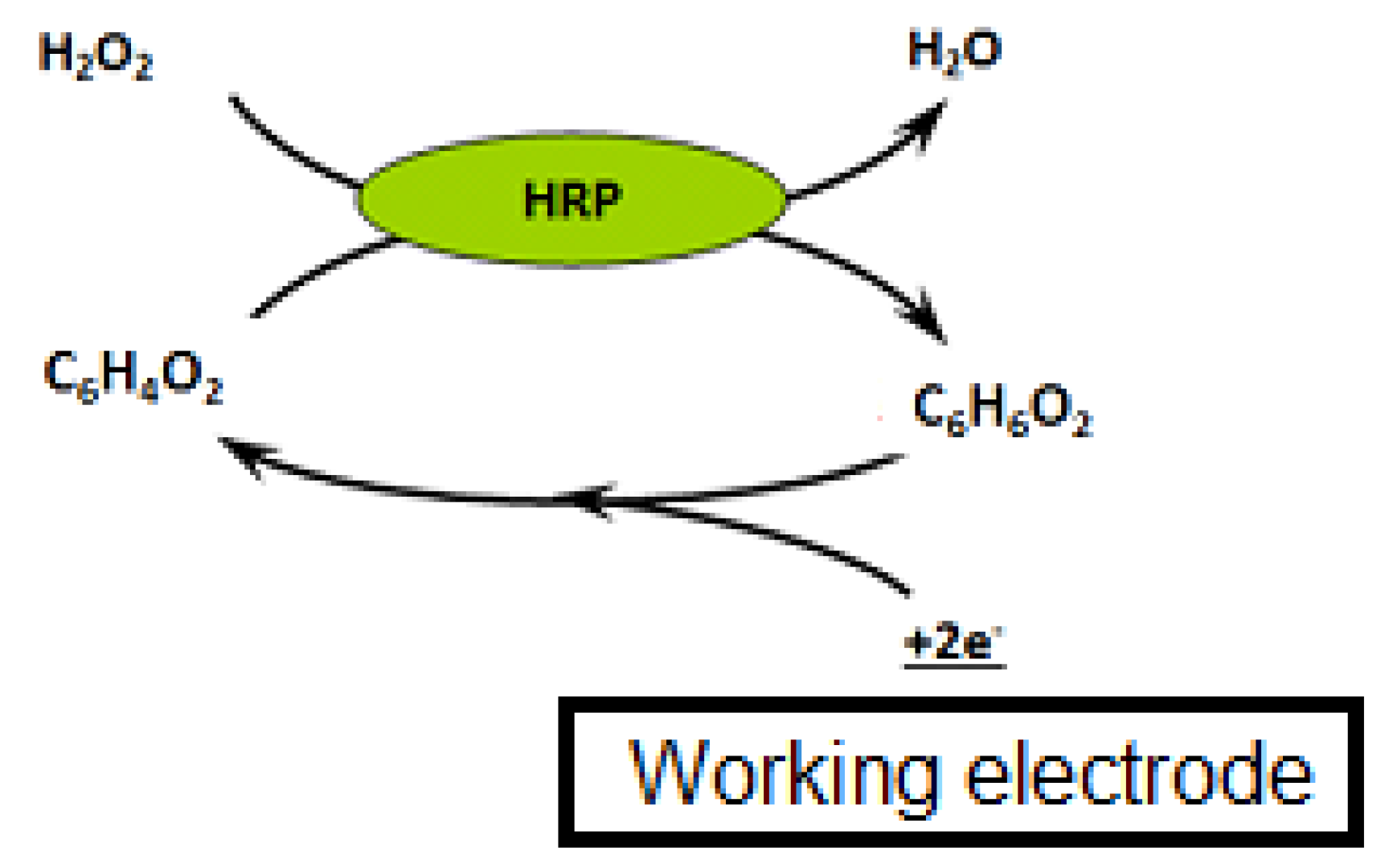
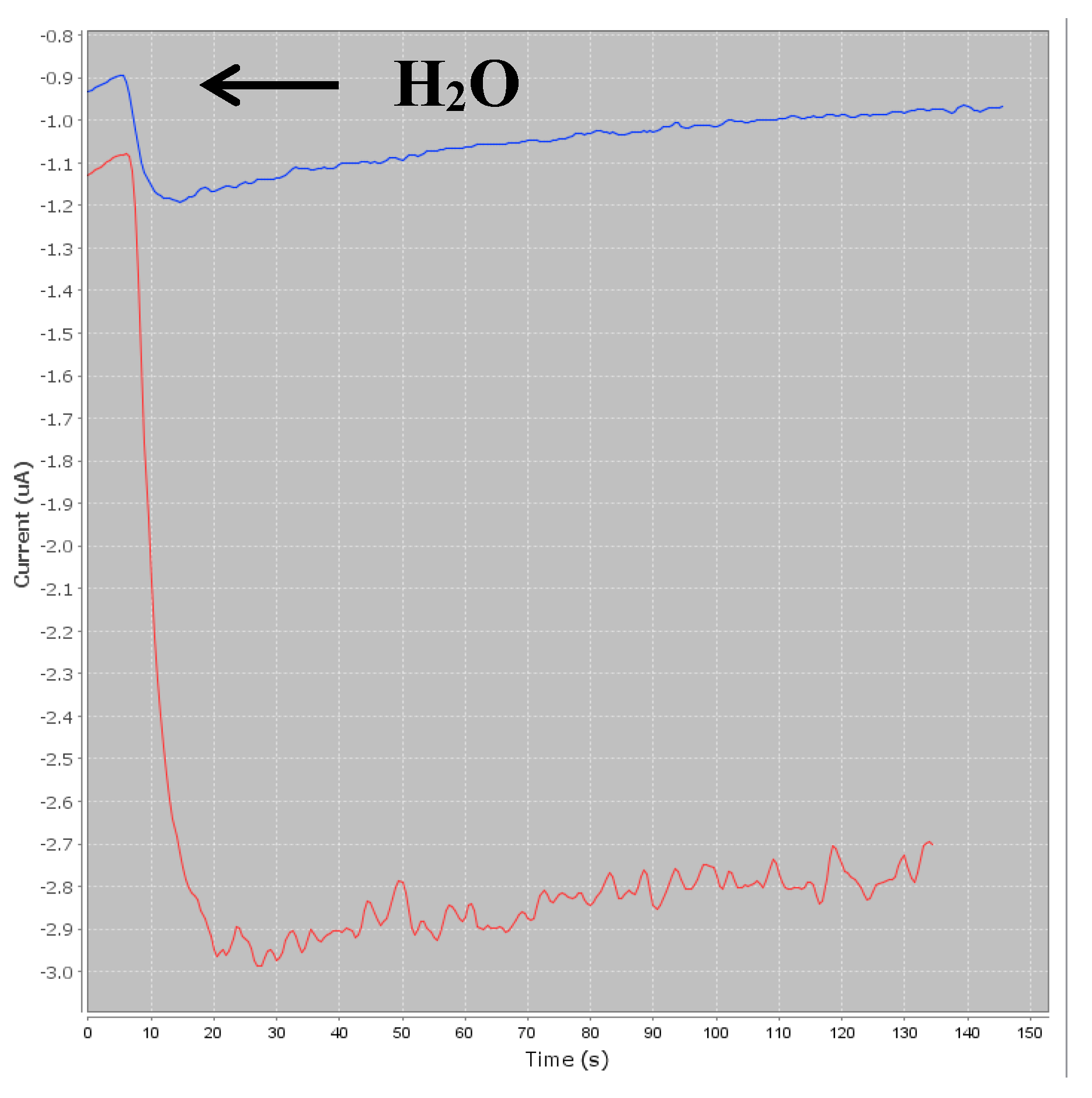
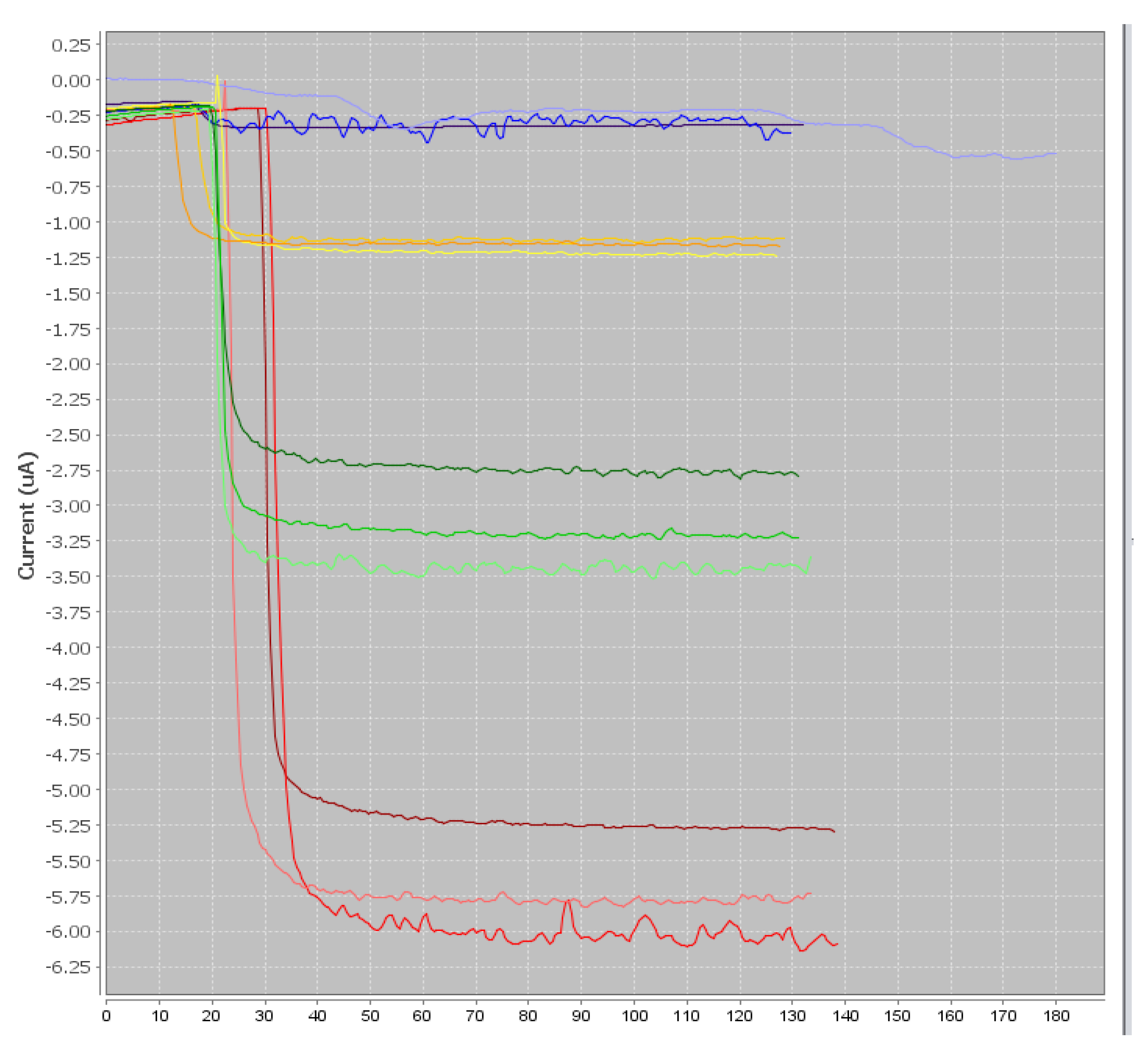
Three microliters of beads were rinsed twice with PBS to remove the sodium azide preservative, then the supernatant was separated from the beads using a magnetic support that retains the beads on the walls of the tubes. These beads were then placed in contact with 50 µL of antibodies. The antibodies were grafted in a thermoshaker for 30 min at 37 °C and at 1200 rpm. Then, the beads were washed twice in PBS-T to remove any unbound antibodies. Fifty microliters of a mix of PBS-T (or a CAP solution or honey sample) with CAP–HRP conjugate were added to the tube. While the incubation was taking place, a solution of hydroquinone (275 mg to 25 mL phosphate buffer) was prepared in phosphate buffer, as well as a solution of hydrogen peroxide (520 µL to 5 mL of phosphate buffer). After incubation, the beads were rinsed twice with PBS-T (100 µL) to remove compounds in excess that were not attached to the antibodies and the honey extract. Pending reading of the results, the beads were set aside in 50 µL of phosphate buffer. Then, 50 µL of magnetic beads were deposited on the electrode, which itself was attached to a magnetic support, which was plunged into an electrochemical cell containing 12 mL of phosphate buffer and 1300 µL of hydroquinone solution. Hydroquinone acts as an electron transducer to facilitate the redox reaction of hydrogen peroxide catalyzed by HRP. Electrons will be taken up by the hydroquinone, and therefore be taken up by the electrode, and this signal will be converted to a readable signal by the computer (
Figure 2). A known potential (−0.25 V) was applied to the electrode and when a well-stabilized baseline appeared on the screen, 65 µL of H
2O
2 solution was added (
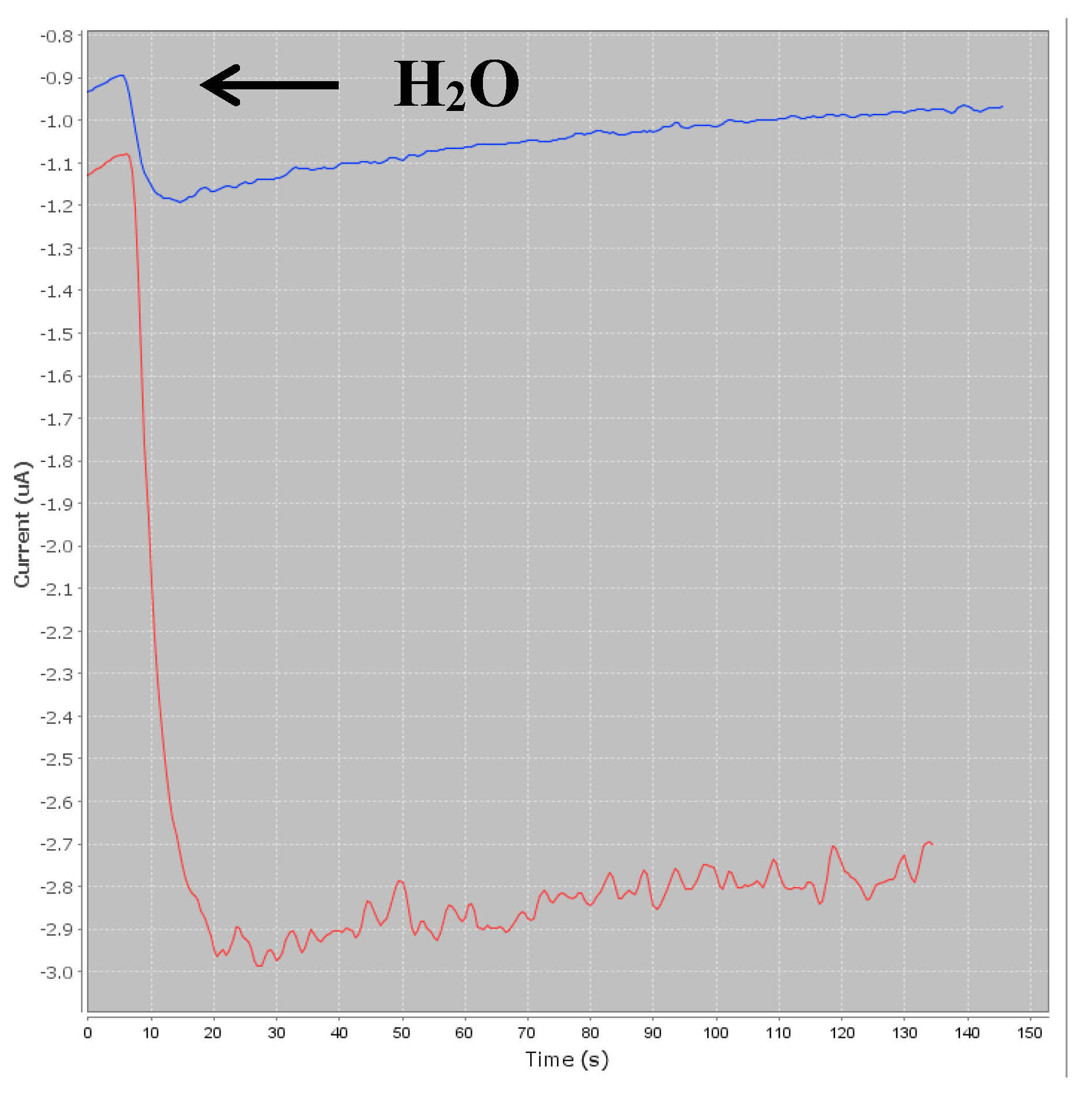
Figure 3).
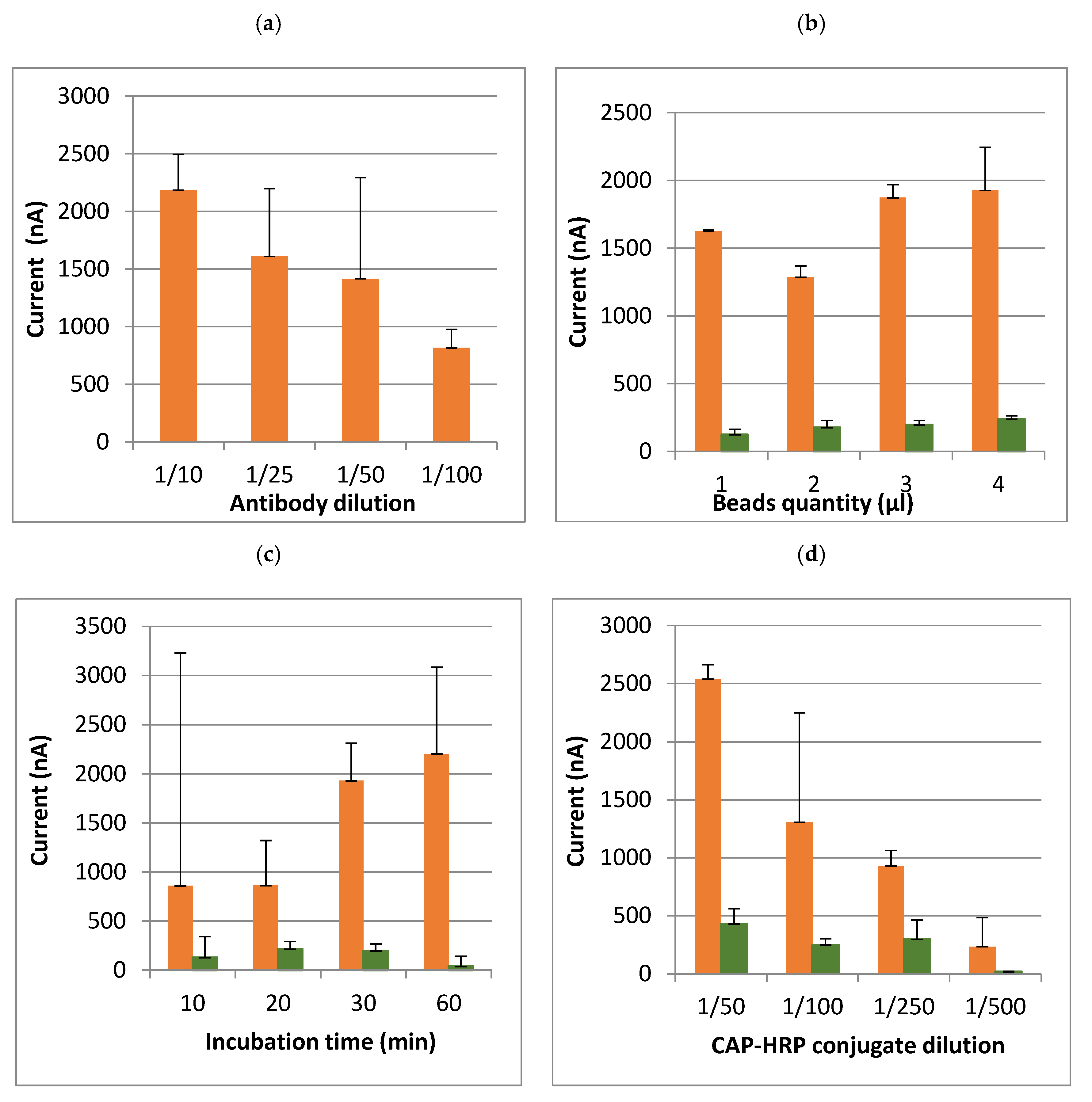
2.7. Optimization of Assay Conditions
Before starting the optimization, the first test was carried out with a moderate quantity of beads (5 µL) and a high concentration of antibodies and conjugate in PBS-T. Then, the assay conditions were optimized in buffer with a CAP concentration of 10 ng/mL.
The aim of the optimization was to determine the technical conditions that were a compromise between sensitivity (limit of detection, at least less than or equal to the regulatory limit), and cost of analysis. Several steps in the method have been optimized (
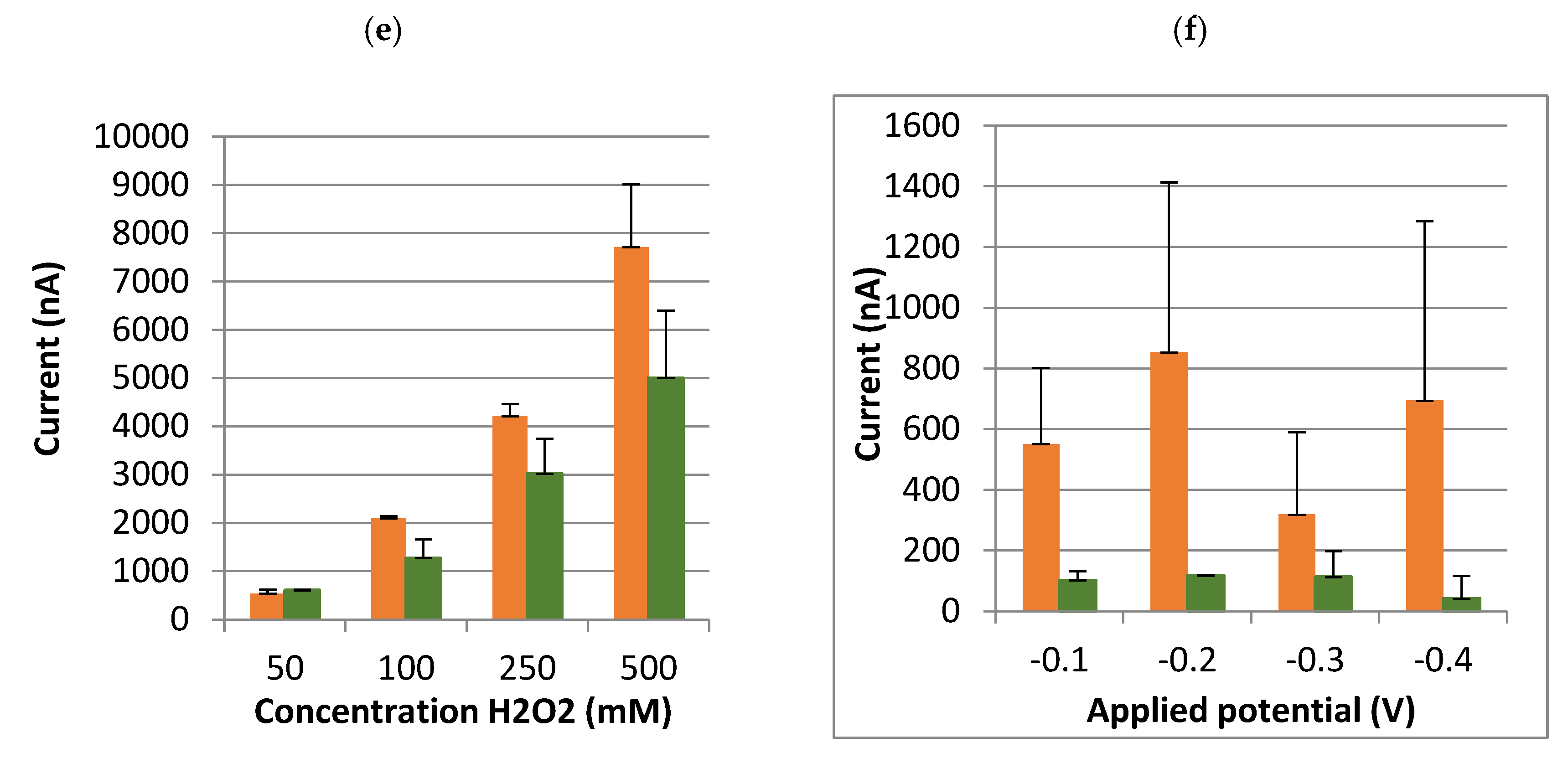
Table 1): quantity of magnetic beads, temperature, time and intensity of rotation for the antibody incubation with the beads, CAP–HRP conjugate, hydroquinone and hydrogen peroxide concentrations, and incubation methods (i.e., a single incubation or a sequential incubation). Lastly, it is possible to adjust the potential applied by the potentiometer (between −0.1 and −0.4 V).
2.9. Data Analysis
The intensity of the current was measured. For each day of analysis, the mean, standard deviation (SD) and coefficient of variation (CV%) were calculated for a set of identical samples (blank or spiked samples). Then, a statistical approach which took into account the β error of 5% was chosen as it was recommended in the European guidelines for the validation of screening methods for veterinary drug residues [
26].
The positivity threshold T (Equation (1)) and the cut-off value Fm (Equation (2)) were calculated.
where B is the mean and SDB is the standard deviation of the signal (Relative Luminescence Unit (RLU)) of the blank samples.
where M is the mean and SD is the standard deviation of the signal (RLU) of the spiked samples.
The assay was considered valid only if T was higher than Fm.
4. Discussion
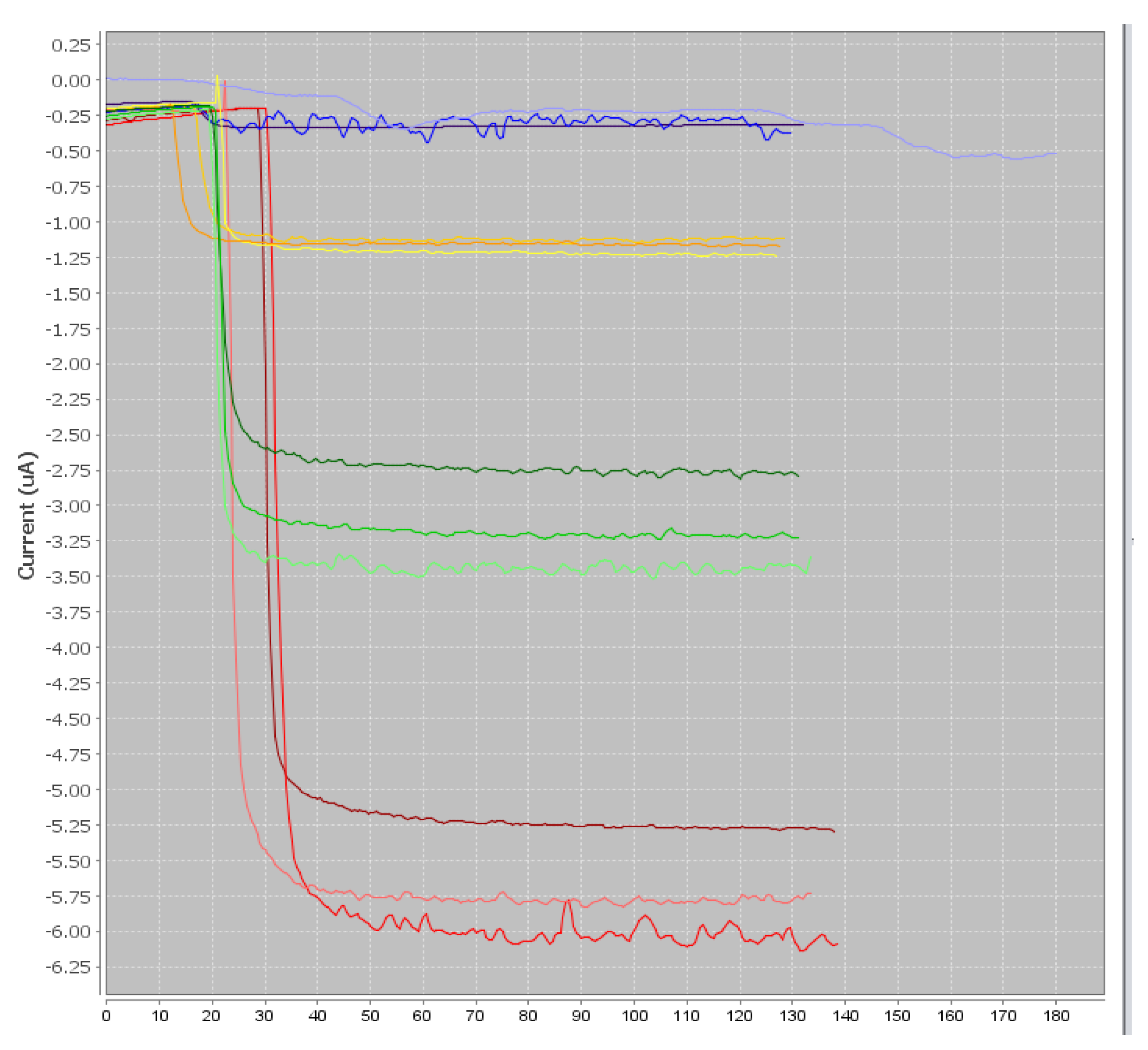
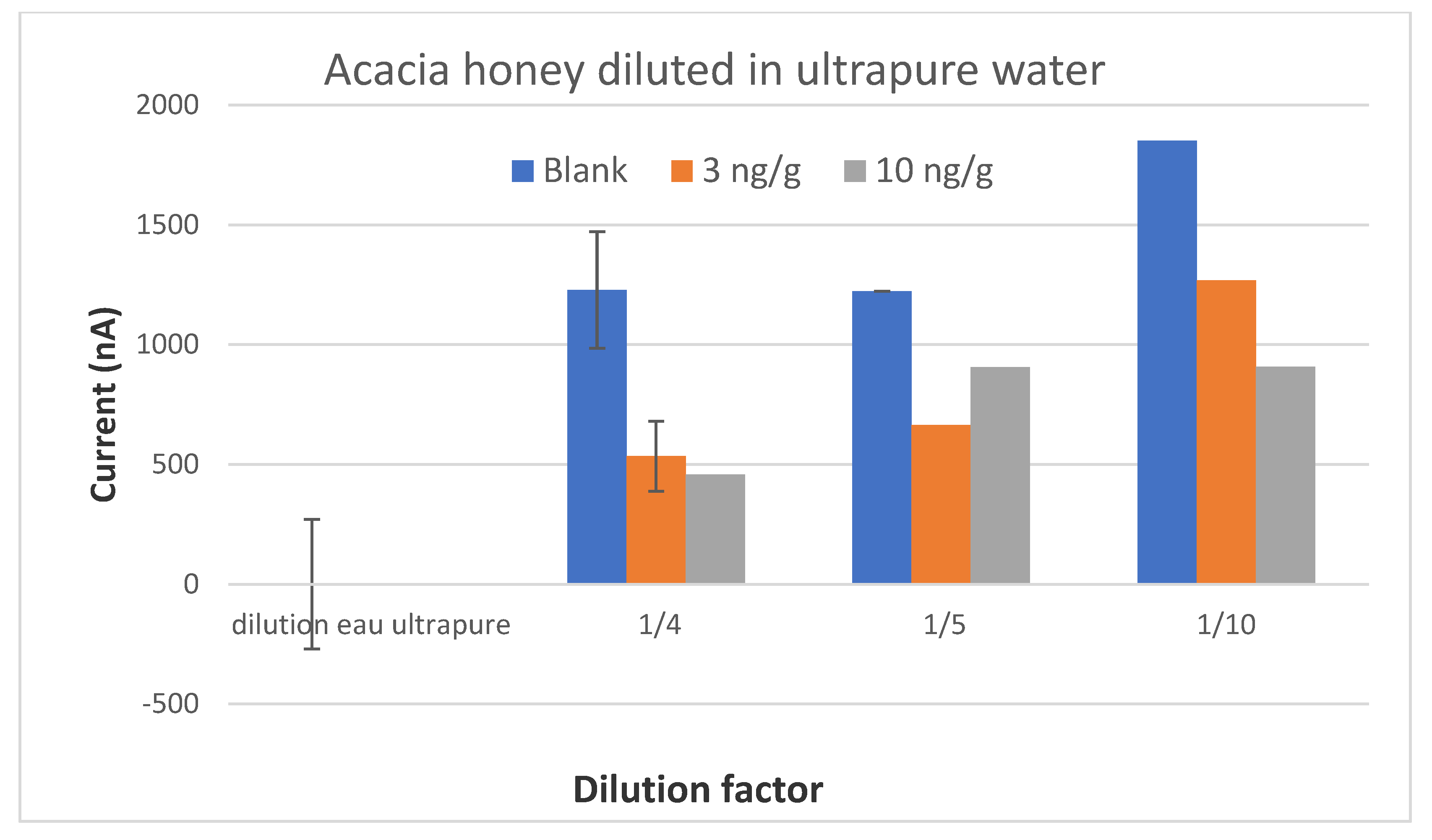
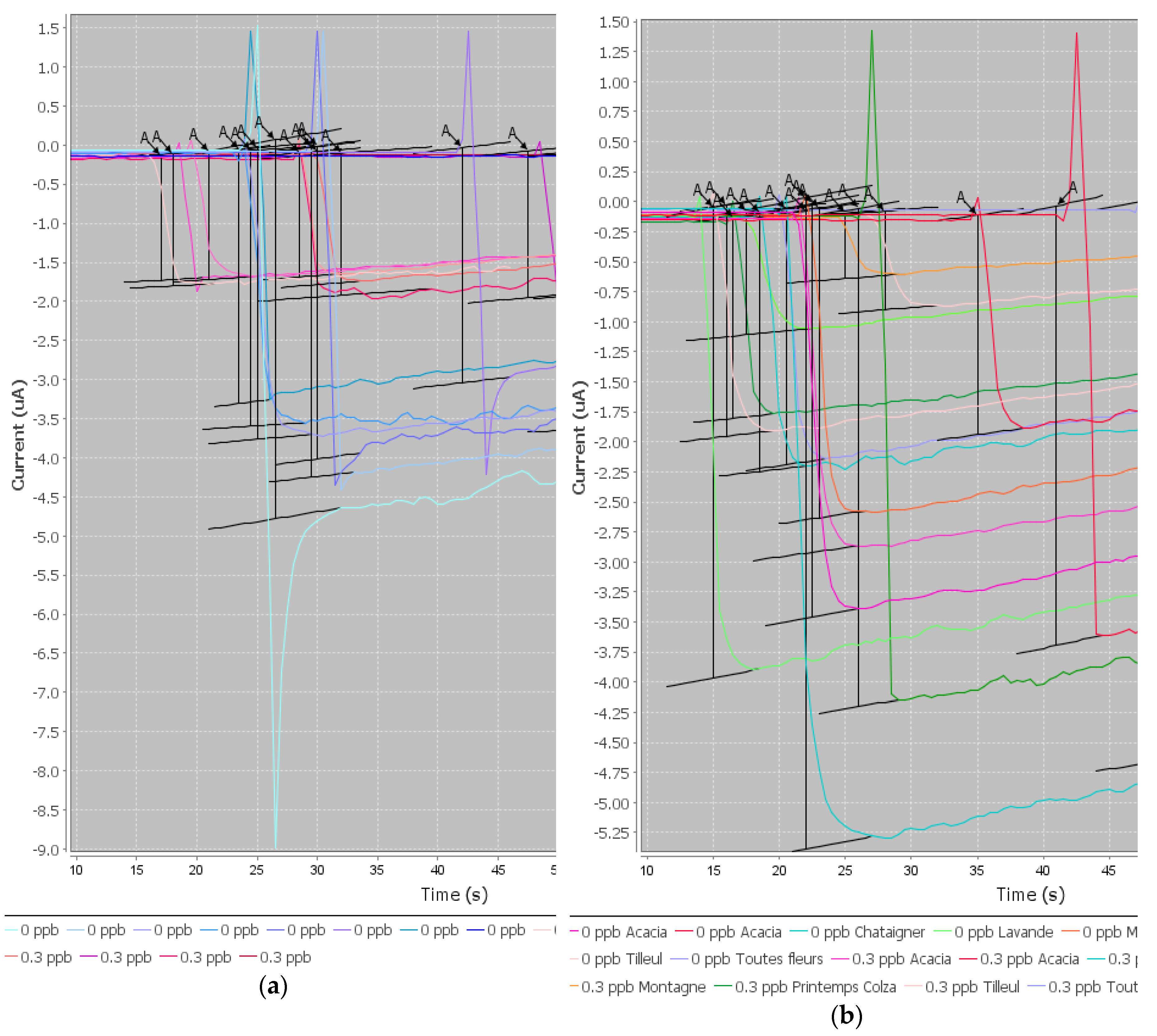
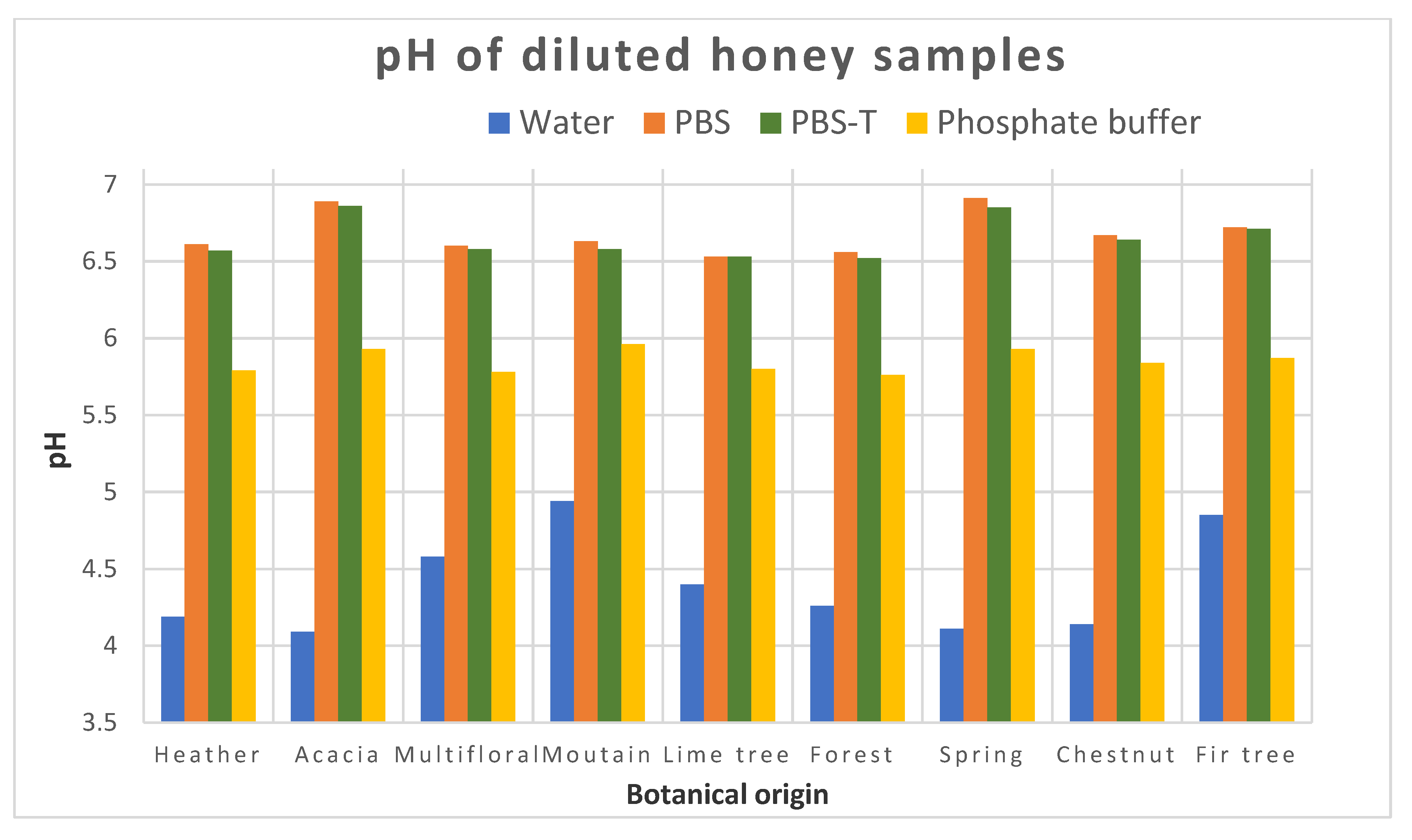
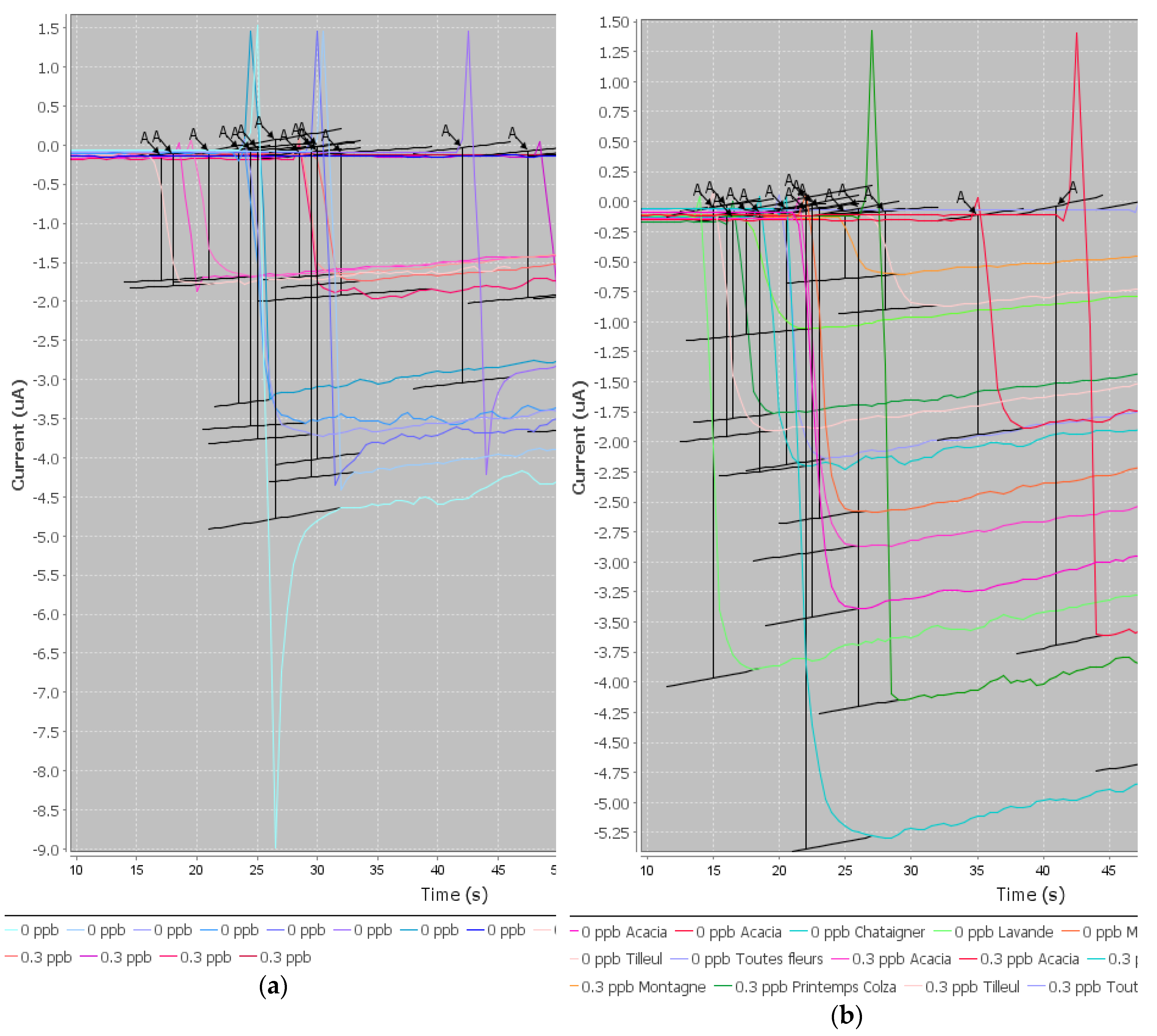
The preliminary results of this project were satisfactory because we were able to develop a screening test able to detect CAP at or below 0.3 ng/g in one honey sample. When individual honeys were analyzed, the method was able to discriminate blank samples from samples spiked with CAP to 0.3 ng/g. Therefore, this objective is fulfilled. However, as soon as the mean signals from honeys of different botanical origins were considered, it was no longer possible to discriminate blank samples and spiked samples because the variability in the signal due to each honey was too high.
After an exhaustive literature study, several extraction protocols for CAP in honey were selected and tested. A simple dilution of honey in buffer would have been an ideal sample preparation. However, the results were not satisfactory when honeys from different botanical origins were tested. The extraction of CAP in ethyl acetate is actually a very usual procedure before analysis with ELISA kits or physico-chemical methods. We have tested different protocols with ethyl acetate and also some protocols with acetonitrile. It was not satisfactory as the matrix effect was not removed.
It was hypothesized that some components interfered with the electrochemical detection. Honey is a very complex matrix of many molecules such as sugars, proteins, vitamins, minerals, polyphenols, organic acids, and lipids. The issue is that a number of these compounds are known to act as antioxidants (i.e., polyphenols (flavonoids and non-flavonoids), organic acids, vitamins (ascorbic acid), and enzymes (glucose oxidase and catalase)). Many of the honey flavonoids and phenolic acids are known to have antioxidant activity [
29]. The amount and type of flavonoids vary depending on the flower source (i.e., botanical origin). Honeys from seven different floral sources were analyzed for in vitro antioxidant capacity and total phenolic content by Gheldhof et al. [
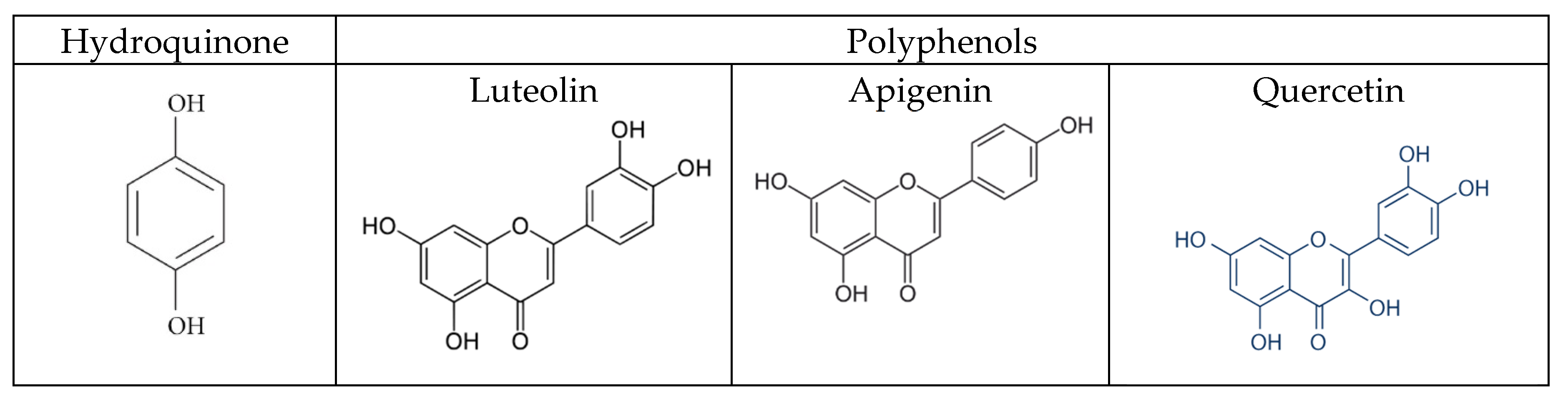
30]. A correlation was observed between phenolic content, honey color and the antioxidant capacity of the investigated honeys. Honeys with a darker color have a higher total phenolic content and consequently a higher antioxidant capacity. Phenols are hydroxylated derivatives of benzene-like hydroquinone. Polyhydroxy derivatives are called polyphenols. The polyphenols present in honey are mainly flavonoids (quercetin, luteolin, apigenin), phenol acids and their derivatives. The darker the honey (like those from sunflower, buckwheat and honeydew), the richer they are in flavonoids. The structure of these flavonoids is very similar to those of hydroquinone (
Figure 9). Furthermore, the oxidation of polyphenols produces quinones. The oxidation of polyphenols could prevent the oxidation of hydroquinone and therefore all the electrochemical reactions that have to be measured.
The difficulty arises with finding a solvent which could extract these substances without extracting CAP. The issue is that phenolic acids and flavonols are usually extracted from honey in ethyl acetate or ethanol, like CAP. Only sugars are removed by this extraction. This co-extraction could explain the strong matrix effect we have observed. Further studies to find a solvent that would extract CAP but not antioxidant components of honey, if possible, are needed. The solubility of flavonoids also depends on the pH [
31]. Many natural flavonoids have extremely low solubility in aqueous media [
32]. A combination of several solvents and the variation of pH for the extraction step should be tested.
One big challenge in the development of biosensors for the detection of analytes is working with biological matrices. This issue of interferences when handling biological samples has already been highlighted in several scientific publications [
33]. The sensitivity of the biosensor could be highly decreased because of matrix compound interference. We faced the same issue. While everything goes well in a buffer or in aqueous solution (e.g., sensitivity, selectivity), the analysis of biological matrices is very complicated, especially when the matrix is as varied as honey. We assumed that some substances in the matrix (i.e., electroactive species) interfered with the electrochemical detection, as this has already been observed in previous works [
34]. Most often, research articles on CAP biosensors are evaluated only in buffer or aqueous solution [
35]. When the method is applied to biological matrices, only one or a few matrix samples are analyzed [
36,
37,
38]. This study is only a proof of concept for the developed biosensor, but it is not a full evaluation of the actual performance of the method and of its routine applicability. A full validation requires the analysis of at least twenty samples from different origins (i.e., honeys from different botanical and geographical origins) for the determination of specificity and detection capabilities [
2,
26].
The objective of a screening method is to be able to detect CAP in any type of honey. In routine analyses, we carry out negative quality controls (QC) (blank samples) and positive QCs (samples spiked with CAP at the detection capability (or limit of detection)) daily. The analysis of these QC samples makes it possible to determine the daily T and Fm. Thus, we can conclude if the test of the day is indeed valid. Finally, the cut-off Fm is used to evaluate the positivity of the unknown samples analyzed on the same day. Therefore, at least two blank honey samples and two spiked honey samples from different botanical origins preferably have to be analyzed. However, if the signals are too different between botanical origins, this management of routine analyses will not be possible. Therefore, we must develop an analytical method, combining sample extraction and analysis on the biosensor, able of analyzing all types of honey simultaneously. Otherwise, two negative and two positive QCs would be necessary for each floral origin to be analyzed on the day of the test, which would be completely impractical.
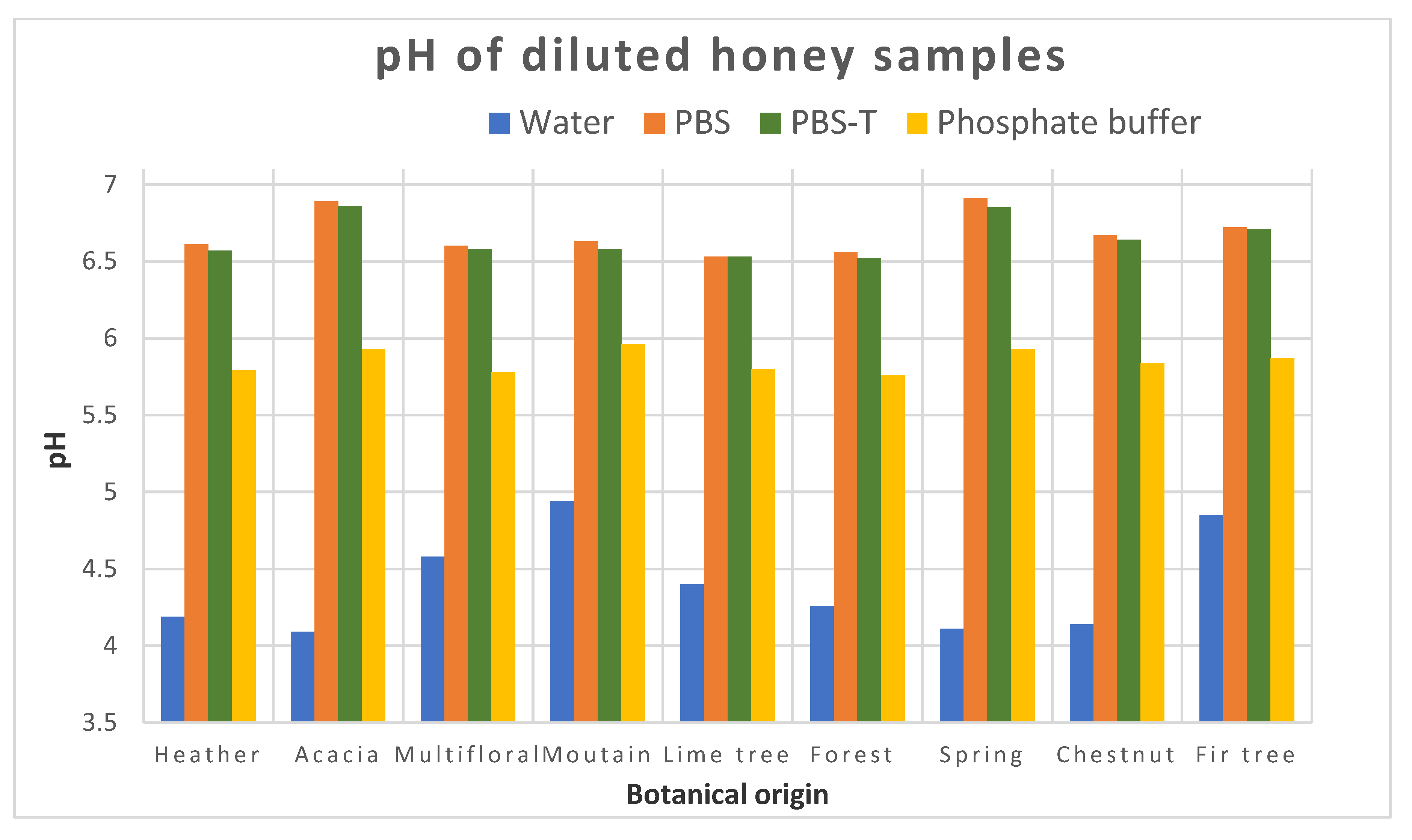
If a common extraction protocol removing matrix effects is not available, we could consider in the future making groups within the variety of honeys. To form these groups, we would need more results to be able to find which types of honey could be integrated in each group. We assumed that the groups could be linked to the color of honey, or to its pH, for instance. This will require analyzing QCs from each group for the analysis of unknown samples based on the botanical origin of the samples to be analyzed.
In conclusion, SPCE are attractive for the development of biosensors for the detection of antimicrobial residues in foodstuffs because they are relatively affordable, disposable (no cleaning process, no memory effect), and can be portable (i.e., small, robust, simple, portable instrumentation) [
33]. Magnetic beads are usually useful because they help to minimize the matrix effect (i.e., blocking of the binding sites on the electrode) due to the washing steps, but they also enable quicker analysis because they are in suspension. In addition, the sensitivity is improved, and the immuno-reaction step is separate from the electrochemical detection step. Unfortunately, regarding the complexity of the honey matrix and the great variety in its components, even the use of magnetic beads did not allow us to remove all matrix effects. For more than ten years, nanomaterials (NMs) and their outstanding characteristics have been widely used in the development of very sensitive electrochemical biosensors [
39,
40]. Nanomaterials improve the sensitivity of biosensors by increasing the surface area of the transduction zone. They can improve the electrical conductivity in the case of electrochemical biosensors and reduce the background noise associated with the samples analyzed (matrix effect). Unfortunately, it seemed that the tested modified SPCE did not give better results than the non-modified SPCE, and even gave worse results for some samples. Further tests in buffer should be conducted to verify if the problem was due to the strong matrix effect of the honey.
There are many perspectives to this work:
We will continue to develop more efficient extraction protocols that are able to remove or at least lower the strong matrix effect of honey.
The positive effect of PVPP on the reduction of the matrix effect will continue to be investigated, probably firstly by increasing its concentration.
We will compare different electrochemical modes of detection, such as amperometry and voltammetry (i.e., voltamperometry).
The use of nanomaterials could be explored to improve the performance characteristics of the electrode (i.e., modified SPCE).
An aptamer will be evaluated in replacement of the antibody to detect CAP. Aptamers are already easily found commercially because the sequence is known [
41]; many articles are working with aptasensors for CAP [
39,
42].
After chloramphenicol, we will develop biosensors for other banned antimicrobials (i.e., nitrofuran metabolites) and finally develop a multiplex method for the simultaneous detection of these banned substances.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}