Abstract
We investigate the dielectric properties of homovalent (M4+)-doped Ba(Ti1−xMx)O3 compositions using a two-dimensional Ising-like network. The model is mainly based on the interaction of permanent and induced dipoles and allows us to simulate the collective behavior of atoms at finite temperatures. In contrast to previous publications, we also include first-principles calculations to model the local environment and interaction of the B-site atoms. Furthermore, in order to describe the corresponding physics more accurately, we introduce an additional degree of freedom for the polarization direction. Our simulations provide an insight into the formation of polar clusters, the evolution of spontaneous polarization at different concentrations of dopants, and the response to external fields. For the purpose of studying the dielectric properties, the model is used to calculate hysteresis curves and related quantities.
1. Introduction
The progress towards energy-independent, autonomous devices is primarily limited by the availability of suitable storage systems. In general, the requirements for those systems are a high energy and power density. The former gives credit to the amount of energy stored per unit mass, whereas the latter is the ability to charge and discharge very fast. A promising type of such energy storage device can be found in dielectric capacitors on the condition that the dielectric fulfills specific requirements. For example, a high permittivity, a small remnant polarization, small leakage currents and a high dielectric breakdown strength. Relaxor ferroelectrics (RF) are one kind of material that are able to meet many, if not all, of the aforementioned requirements. In our research, we focus on barium titanate, which shows RF behavior (and highly promising properties for application in high-energy density storage applications) provided that it is doped with proper atoms and concentrations. In order to avoid synthesizing thousands of different compositions to find the most suitable one, we support our search with Monte Carlo simulations. That allows us to screen through a lot of different compositions with rather less effort, simultaneously studying the microscopic behavior of our materials.
2. Methodology
In our simulations, we use an adapted version of the Ising-like network [1,2]. The main idea is to use permanent and induced dipoles to model the microscopic behavior. The permanent dipoles are related to the displacement of the B-site atoms. To parametrize that, we calculate the potential energy surface (PES) of the local soft mode via density functional theory (DFT) calculations. From that, we extract the averaged displacement by considering the local minima in the PES. Furthermore, the oxygens are moved in those calculations according to the soft mode eigenvector. The corresponding displacements from their equilibrium positions are considered as induced dipoles consisting of two contributions, i.e., atomic and ionic polarizability. By using these definitions, one can easily calculate the local electric field at any unit cell, as stated in Equation (1).
This electric field influences the shape of the local PES and, therefore, is the driving force for a reorientation or stabilization of a specific orientation of permanent dipoles. The induced dipoles arrange according to the configuration of the permanent dipoles and, again, contribute to stabilization or destabilization. To simulate such a system at finite temperatures, a Monte Carlo (MC) algorithm is applied to update the configurations and to converge the system to an equilibrium state. From that, several statistical values are calculated and recorded during the simulation.
3. Results
As illustrative example for the suitability of our simulations, hysteresis loops at different concentrations of Zr in Ba(Ti1−xZrx)O3 are presented in Figure 1. The graph shows the decrease in remnant polarization (which is beneficial to high energy density) by increasing the amount of Zr in the system. This is accompanied by the decline in maximum polarization, which in turn reduces the energy density. Thus, it is clear that an optimum concentration must be found where energy density is maximized.
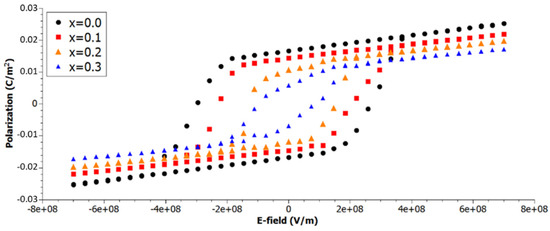
Figure 1.
P(E) loops of Ba(Ti1−xZrx)O3 at different concentrations x of Zr. The temperature during the simulation was fixed at 200 K, whereby the number of MC-sweeps per E-field step was set to 2000. The Zr atoms were randomly distributed on a two-dimensional grid of size 16 × 16.
4. Discussion
The results obtained from our MC simulations of Zr-doped BaTiO3 are in good agreement with comparable values found in the literature [2]. Of note, a quantitative comparison to experimental values is currently not possible due to the reduced dimensionality and the neglect of short-range interaction.
Funding
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No 817190).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Nayel, F.; Herbert, K. Ferroelectricity in systems of induced and permanent dipoles: A Monte Carlo study. J. Appl. Phys. 2001, 90, 5713. [Google Scholar]
- Leontin, P.; Cristian, E.; Liliana, M. Monte Carlo simulations for describing the ferroelectric-relaxor crossover in BaTiO3-based solid solutions. J. Phys. Condens. Matter 2011, 23, 325901. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).