


Development of a Predictive Model for the Biological Activity of Food and Microbial Metabolites Toward Estrogen Receptor Alpha (ERα) Using Machine Learning

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
- 1.
- Physicochemical PropertiesThis category includes descriptors characterizing the basic physicochemical parameters that affect the behavior of molecules in biological systems:
- MolWt (molecular weight): the total mass of the molecule, influencing pharmacokinetic parameters such as distribution and excretion;
- ExactMolWt (exact molecular weight): a more precise mass value taking into account the isotopic composition of atoms, important for mass spectrometric analysis;
- MolLogP: the logarithm of the octanol/water partition coefficient, reflecting the lipophilicity of the molecule and its ability to penetrate biological membranes;
- MolMR (molar refractivity): a parameter related to the polarizability and volume of the molecule, influencing interactions with electromagnetic radiation;
- TPSA (topological polar surface area): the total area of polar atoms and functional groups, correlating with cell membrane permeability and bioavailability.
- 2.
- Structural PropertiesThese descriptors characterize structural features of the molecules:
- HeavyAtomCount: the number of atoms excluding hydrogen, reflecting the molecule’s size and complexity;
- NumValenceElectrons: the total number of valence electrons across all atoms, influencing the chemical reactivity of the molecule;
- NumHAcceptors: the number of atoms (commonly oxygen and nitrogen) capable of accepting hydrogen bonds;
- NumHDonors: the number of groups (usually –OH and –NH) capable of donating hydrogen bonds;
- NumRotatableBonds: the number of single bonds not in rings, defining the molecule’s flexibility;
- RingCount: the total number of cyclic structures in the molecule, influencing its rigidity and spatial configuration;
- FractionCSP3: the proportion of sp3-hybridized carbon atoms, indicating the degree of saturation and three-dimensional character of the molecule;
- AromaticProportion: the ratio of aromatic atoms to the total number of atoms, important for π–π interactions.
- 3.
- Surface and Volume PropertiesThese descriptors evaluate the molecular surface and volume:
- LabuteASA (Labute’s approximate surface area): an estimate of the molecule’s surface area accessible for solvent interaction, affecting solubility;
- PEOE_VSA1–PEOE_VSA10: descriptors of surface area weighted by partial atomic charges, reflecting the distribution of electron density on the molecular surface;
- SlogP_VSA1–SlogP_VSA10: descriptors weighted by the log of the partition coefficient (LogP), characterizing the molecule’s lipophilic and hydrophilic regions;
- SMR_VSA1–SMR_VSA10: descriptors weighted by molar refractivity, reflecting the molecule’s volumetric and polarizable regions;
- EState_VSA1–EState_VSA10: descriptors weighted by electron-topological states of atoms, accounting for electronic effects and molecular topology.
- 4.
- Topological IndicesTopological indices characterize the molecular structure in terms of shape, size, and complexity:
- Chi0 and Chi1: connectivity indices of the 0th and 1st orders, indicating the degree of branching;
- Chi0n–Chi4n: normalized connectivity indices that account for variations in atomic properties and connectivity at different levels;
- Chi0v–Chi4v: connectivity indices weighted by the valence of atoms, reflecting the molecule’s electronic structure;
- Kappa1, Kappa2, and Kappa3 (Kier shape indices): parameters describing the shape and flexibility of a molecule based on its graph structure;
- BalabanJ (Balaban index): a topological index incorporating information on both cyclicity and branching [26];
- BertzCT (Bertz complexity index): a quantitative measure of molecular structural complexity;
- Ipc (information content index): an information-based metric derived from molecular symmetry and bond diversity;
- HallKierAlpha: a parameter related to the correction of atomic properties affecting hydrophobicity and molecular flexibility.
- 5.
- Counts of Specific Atoms and GroupsThis category includes descriptors that account for the presence of certain atoms and functional groups:
- NHOHCount: the total number of hydroxyl (–OH) and amino (–NH) groups, influencing the molecule’s hydrophilicity and reactivity;
- NOCount: the total number of nitrogen and oxygen atoms, reflecting the potential for hydrogen bonding.
- 6.
- Target Variable
- Activity: A binary variable indicating the known biological activity of a compound with respect to estrogen receptor alpha (ERα). It serves as the target variable for training the machine learning model.
- Features (X): a set of molecular descriptors computed for each compound;
- Labels (y): a binary variable (Activity) indicating whether a compound is active (1) or inactive (0).
- Training set (X_train, y_train): used for training and simultaneously optimizing all models;
- Test set (X_test, y_test): used for the final assessment of the already-optimized models on “unseen” data.
- Random forests: RandomForestClassifier (with and without class weighting), BalancedRandomForestClassifier;
- Ensemble methods: AdaBoostClassifier, GradientBoostingClassifier, BaggingClassifier, EasyEnsembleClassifier;
- Linear models: LogisticRegression (with and without class weighting), RidgeClassifier;
- Support vector methods: SVC, LinearSVC;
- Decision trees: DecisionTreeClassifier (with and without class weighting), ExtraTreeClassifier;
- Nearest neighbors: KNeighborsClassifier;
- Naive Bayes classifiers: GaussianNB, BernoulliNB;
- Discriminant analysis: LinearDiscriminantAnalysis, QuadraticDiscriminantAnalysis;
- Neural networks: MLPClassifier;
- Other models: SGDClassifier, GaussianProcessClassifier, VotingClassifier, CalibratedClassifierCV;
- Boosting methods: XGBoostClassifier, CatBoostClassifier (with and without class weighting).
- Hyperparameter Tuning
- 2.
- k-Fold Cross-Validation
- 3.
- Search Methods
- 4.
- Class Balance Adjustment
- 5.
- Selecting Optimal Configurations
3. Results
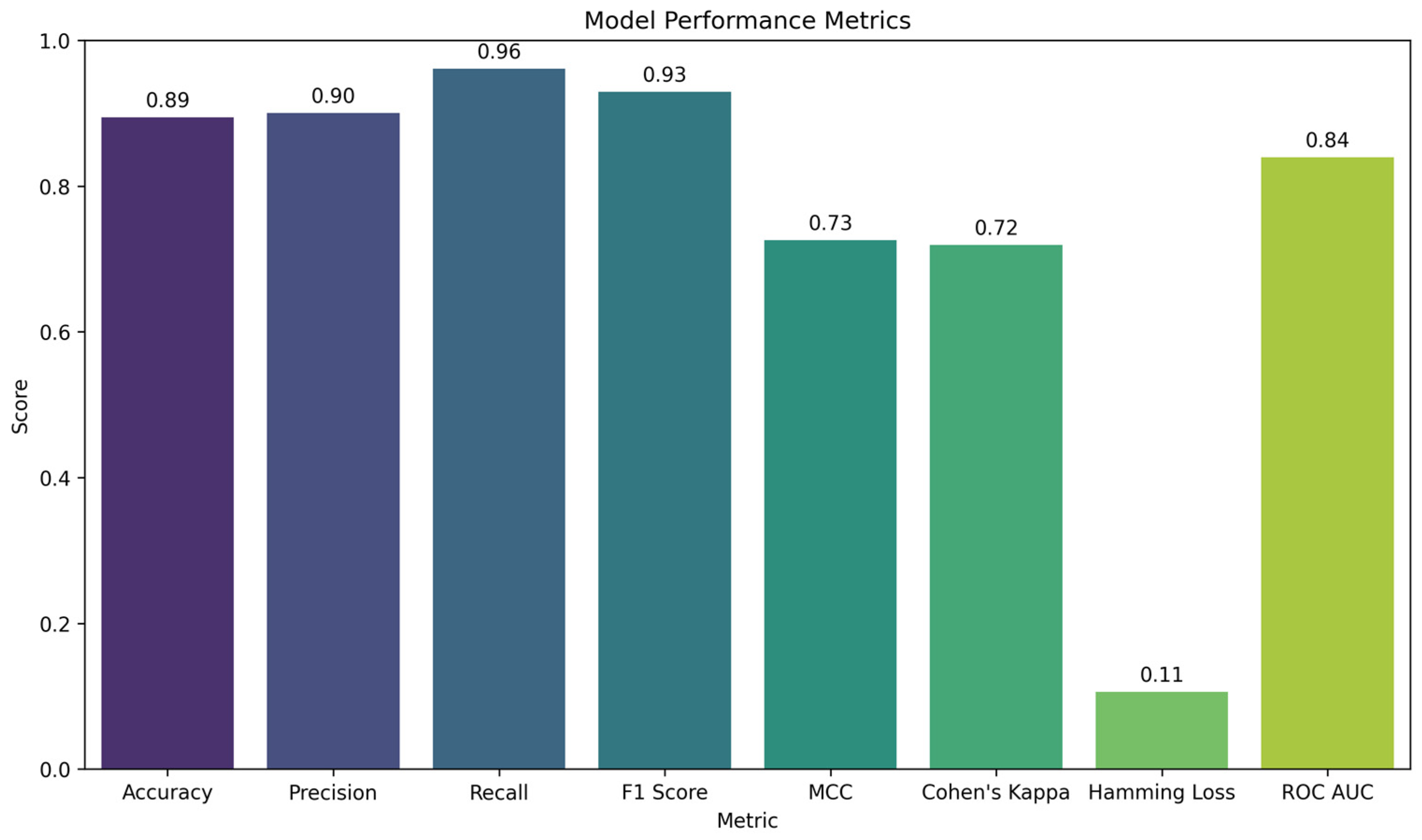
- Accuracy: The proportion of correct predictions among all predictions. A value of 89.44% indicates high overall model accuracy;
- Precision: The proportion of true positives among all predicted positives. A high value (90.00%) signifies a low rate of false positives;
- Recall: The proportion of true positives among all actual positive instances. A value of 96.12% indicates that the model effectively detects active compounds;
- F1 score: The harmonic mean of Precision and Recall. A value of 92.96% reflects a good balance between them;
- MCC (Matthews correlation coefficient): The correlation between observed and predicted classes, accounting for true/false positives and negatives. A value of 0.73 points to a strong positive correlation;
- Cohen’s kappa: A measure of agreement between the model’s predictions and the actual labels, adjusted for chance. A value of 0.72 indicates good agreement;
- Hamming loss: The proportion of incorrect labels relative to the total number of labels. A low value of 0.11 indicates few classification errors;
- ROC AUC score: The area under the receiver operating characteristic curve, shows the model’s ability to discriminate between classes. A value of 83.96% demonstrates a good discriminatory capacity.
- LinearDiscriminantAnalysis (LDA) achieved the highest accuracy (≈0.8944). It also demonstrated high Recall (≈0.9612) and balanced Precision (≈0.9000), leading to an F1 Score of ≈0.9296. LDA further exhibited comparatively high MCC (≈0.7255) and Cohen’s Kappa (≈0.7192);
- RidgeClassifier (Accuracy ≈ 0.8873) slightly lags behind LDA but also provides a high Recall (≈0.9515);
- LogisticRegression offers a high Recall (≈0.9903), though its overall Accuracy (≈0.8662) is lower than that of the top models;
- Ensemble methods (Bagging, BalancedRandomForest, GradientBoosting, etc.) generally yield Accuracy values in the 0.80–0.85 range, illustrating the benefits of combining multiple base models;
- Models with adjusted class weights (BalancedRandomForest, CatBoost with class weighting) indeed handle the minority class more effectively, as demonstrated by their Precision/Recall values [32];
- Certain algorithms (SGDClassifier, SVC, LinearSVC, MLPClassifier, etc.) converged on a “hard” boundary (Recall = 1.0 and Precision ≈ 0.725), indicating peculiarities in how they optimize given this dataset;
- GaussianProcessClassifier (Accuracy ≈ 0.3732) displayed the lowest results, likely due to the specific characteristics of this algorithm and the complexity of the data structure.
- High Accuracy (≈0.8944)—the top result among all the models tested;
- Balanced Metrics (Precision ≈ 0.900, Recall ≈ 0.961)—LDA effectively predicts both active and inactive compounds;
- High MCC (0.7255) and Cohen’s kappa (0.7192)—indicate strong agreement between predictions and actual labels, along with a low probability of random alignment.
- By simultaneously tuning hyperparameters across all the models using cross-validation, we ensured an objective and comprehensive comparison of various machine learning algorithms;
- LDA achieved the highest accuracy and exhibited balanced performance across additional metrics, confirming its suitability for this application;
- Models incorporating class weighting demonstrated enhanced sensitivity to the minority class—a critical consideration when dealing with small but biologically significant groups of compounds [32];
- The optimized LDA model is not only effective for screening novel metabolites but also holds promise as a tool for the early stages of drug development, where rapid and reliable evaluation of compound activity is essential.
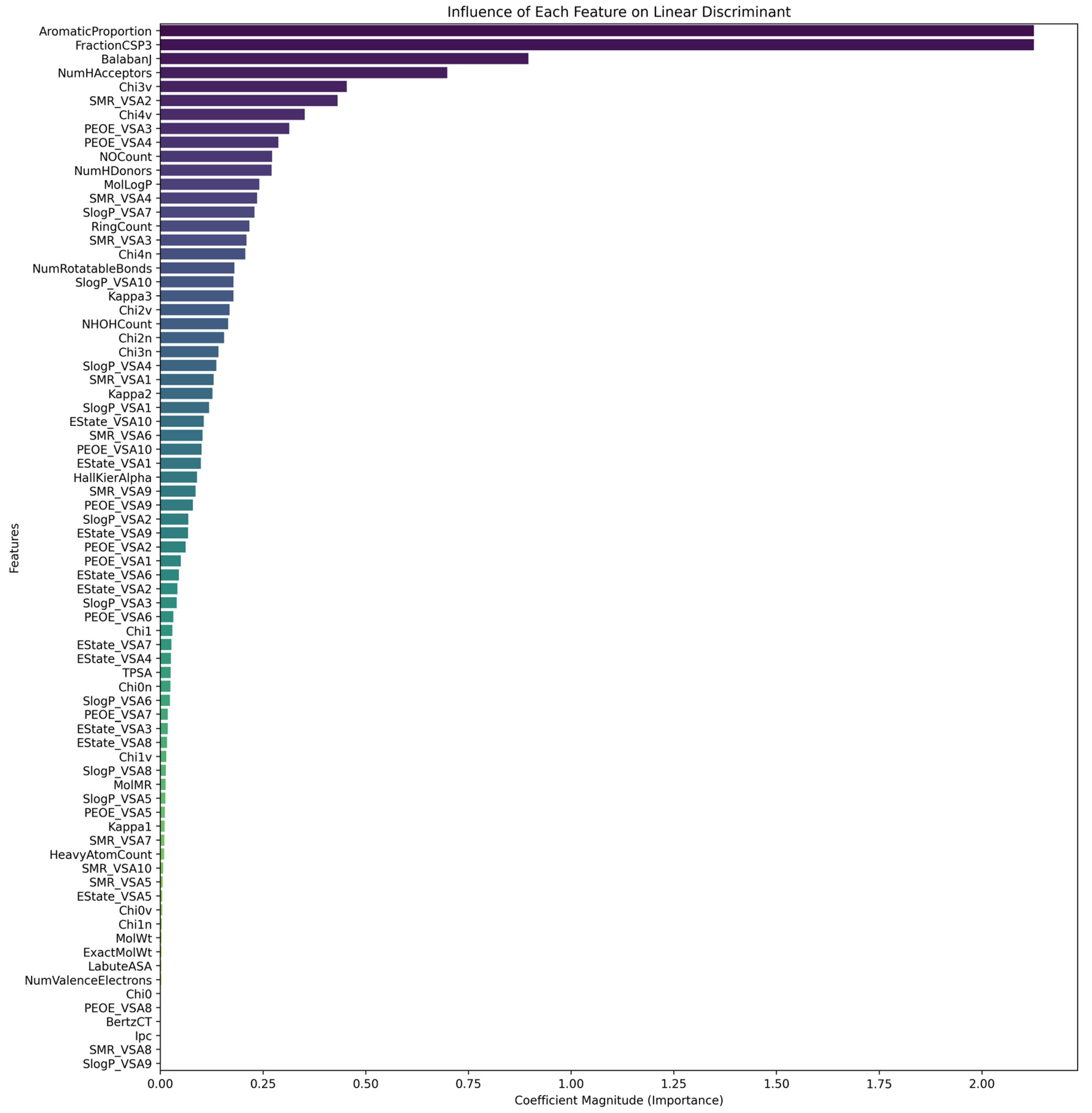
- FractionCSP3 (Proportion of sp3-Hybridized Carbon Atoms)
- 2.
- AromaticProportion (Proportion of Aromatic Atoms)
- 3.
- BalabanJ (Balaban Index)
- 4.
- NumHAcceptors (Number of Hydrogen Bond Acceptors)
- 5.
- Chi3v and Chi4v (Valence-Weighted Connectivity Indices)
- 6.
- SMR_VSA2 (Surface Area Descriptor Weighted by Molar Refractivity)
- 7.
- PEOE_VSA3 and PEOE_VSA4 (Surface Area Descriptors Weighted by Partial Charges)
- 8.
- NOCount (Total Number of Nitrogen and Oxygen Atoms)
- Degree of saturation and aromaticity: the descriptors FractionCSP3 and AromaticProportion indicate that both saturated and aromatic structures are important for ERα interactions;
- Topological complexity: the indices BalabanJ, Chi3v, and Chi4v underscore the importance of molecular complexity and unique structural configurations in receptor binding;
- Hydrogen-bonding capacity: NumHAcceptors and NOCount emphasize the role of hydrogen bonding in stabilizing the ligand–receptor complex;
- Electronic and surface properties: the PEOE_VSA and SMR_VSA descriptors highlight the effects of electron density distribution and surface polarizability on molecular interactions with ERα.
- Guidance in synthesizing new compounds—by focusing on structural features that enhance the likelihood of ERα binding, one can optimize the design of new ligands;
- Improvement of predictive models—considering influential features during model training and potentially removing less informative descriptors can enhance predictive performance;
- Advancement of mechanistic understanding—gaining deeper insight into the structural elements that promote biological activity supports the rational design of more effective therapeutic agents.
- Presence in Food Products:
- 2.
- Influence on Organoleptic Properties:
- 3.
- Potential Biological Activity:
4. Discussion
- Indole Compounds
- Indole and skatole (3-methylindole)
- Indole-3-acetic acid (IAA)
- 2.
- Phenolic Compounds
- 4-Cresol, o-cresol, 3-methylphenol (m-cresol), and phenol
- 3.
- Urolithins
- Urolithin A and urolithin B
- 4.
- Trimethylamine (TMA) and Trimethylamine N-oxide (TMAO)
- TMA and TMAO are found in seafood, especially fish and shellfish. TMAO acts as a natural osmolyte in marine organisms. TMA is responsible for the characteristic fishy odor, which is intensified in spoiled products due to the breakdown of TMAO into TMA. Their activity toward ERα may be relevant for food technologies involving seafood [49].
- Development of technical guidelines that account for processing parameters influencing the formation of active metabolites;
- Use of predictive models, incorporating data on predicted metabolite activity in food quality and safety control systems;
- Professional training of food technologists and quality specialists in microbiological process management and the impact of metabolites on food products.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| LDA | Linear discriminant analysis |
| SVC | Support vector classification |
| ERα | Estrogen receptor alpha |
| TMA | Trimethylamine |
| TMAO | Trimethylamine N-oxide |
Appendix A

{kind=link}
{kind=link}
| No. | Metabolites | Smiles |
|---|---|---|
| 1 | Indole | C1=CC=C2C(=C1)C=CN2 |
| 2 | Skatole (3-methylindole) | CC1=CNC2=CC=CC=C12 |
| 3 | Trimethylamine (TMA) | CN(C)C |
| 4 | Indole-3-acetic acid (IAA) | C1=CC=C2C(=C1)C(=CN2)CC(=O)O |
| 5 | Indole-3-lactate | C1=CC=C2C(=C1)C(=CN2)C(CO)C(=O)O |
| 6 | p-Cresol (4-cresol) | CC1=CC=C(C=C1)O |
| 7 | p-Cresol sulfate | CC1=CC=C(C=C1)OS(=O)(=O)O |
| 8 | Phenylacetic acid | C1=CC=C(C=C1)CC(=O)O |
| 9 | Phenyllactic acid | C1=CC=C(C=C1)C(CO)C(=O)O |
| 10 | o-Cresol | CC1=CC=CC=C1O |
| 11 | 3-Methylphenol (m-cresol) | CC1=CC(=CC=C1)O |
| 12 | Deoxycholic acid | C[C@H](CCC(=O)O)C1CCC2C(C1)CCC3C2CC(C4C3(CCC(C4)O)C)O |
| 13 | Lithocholic acid | C[C@H](CCC(=O)O)C1CCC2C(C1)CCC3C2CC=C4C3(CCC(C4)O)C |
| 14 | Urolithin A | C1=CC(=C2C(=C1)C(=O)C3=CC=CC=C3C2=O)O |
| 15 | Urolithin B | C1=CC(=C2C(=C1)C(=O)C3=CC=CC=C3C2=O)O |
| 16 | Acetoin | CC(C=O)O |
| 17 | 2,3-Butanediol | CC(CO)CO |
| 18 | Cadaverine | C(CN)CCCN |
| 19 | Putrescine | C(CCN)CN |
| 20 | Trimethylamine N-oxide (TMAO) | C[N+](C)(C)[O-] |
| 21 | Phenol | C1=CC=C(C=C1)O |
References
- Ma, H.; Gollahon, L.S. ERα Mediates Estrogen-Induced Expression of the Breast Cancer Metastasis Suppressor Gene BRMS1. Int. J. Mol. Sci. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lee, J.H.; Zhang, Z.; De La Rosa, R.; Bi, M.; Tan, Y.; Liao, Y.; Hong, J.; Du, B.; Wu, Y.; et al. Enhancer RNAs Mediate Estrogen-Induced Decommissioning of Selective Enhancers by Recruiting ERα and Its Cofactor. Cell Rep. 2020, 31, 107803. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-R.; Zhang, R.; Lian, Z.-X.; Deng, S.-L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef]
- Della Torre, S.; Mitro, N.; Fontana, R.; Gomaraschi, M.; Favari, E.; Recordati, C.; Lolli, F.; Quagliarini, F.; Meda, C.; Ohlsson, C.; et al. An Essential Role for Liver ERα in Coupling Hepatic Metabolism to the Reproductive Cycle. Cell Rep. 2016, 15, 360–371. [Google Scholar] [CrossRef]
- Fu, Q.; Li, T.; Zhang, C.; Ma, X.; Meng, L.; Liu, L.; Shao, K.; Wu, G.; Zhu, X.; Zhao, X. Butyrate mitigates metabolic dysfunctions via the ERα-AMPK pathway in muscle in OVX mice with diet-induced obesity. Cell Commun. Signal. 2023, 21, 95. [Google Scholar] [CrossRef]
- Guo, M.; Cao, X.; Ji, D.; Xiong, H.; Zhang, T.; Wu, Y.; Suo, L.; Pan, M.; Brugger, D.; Chen, Y.; et al. Gut Microbiota and Acylcarnitine Metabolites Connect the Beneficial Association between Estrogen and Lipid Metabolism Disorders in Ovariectomized Mice. Microbiol. Spectr. 2023, 11, e00149-23. [Google Scholar] [CrossRef]
- Qi, X.; Zhao, Y.; Qi, Z.; Hou, S.; Chen, J. Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges. Molecules 2024, 29, 903. [Google Scholar] [CrossRef]
- Björnström, L.; Sjöberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.Å. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Knowlton, A.; Lee, A. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Huggins, R.J.; Greene, G.L. ERα/PR crosstalk is altered in the context of the ERα Y537S mutation and contributes to endocrine therapy-resistant tumor proliferation. NPJ Breast Cancer 2023, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Scabia, V.; Ayyanan, A.; De Martino, F.; Agnoletto, A.; Battista, L.; Laszlo, C.; Treboux, A.; Zaman, K.; Stravodimou, A.; Jallut, D.; et al. Estrogen receptor positive breast cancers have patient specific hormone sensitivities and rely on progesterone receptor. Nat. Commun. 2022, 13, 3127. [Google Scholar] [CrossRef] [PubMed]
- Heath, H.; Mogol, A.N.; Casiano, A.S.; Zuo, Q.; Madak-Erdogan, Z. Targeting Systemic and Gut Microbial Metabolism in ER+ Breast Cancer. Trends Endocrinol. Metab. 2024, 35, 321–330. [Google Scholar] [CrossRef]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 1 October 2023).
- Ortega, M.A.; Alvarez-Mon, M.A.; García-Montero, C.; Fraile-Martinez, O.; Guijarro, L.G.; Lahera, G.; Monserrat, J.; Valls, P.; Mora, F.; Rodríguez-Jiménez, R.; et al. Gut Microbiota Metabolites in Major Depressive Disorder—Deep Insights into Their Pathophysiological Role and Potential Translational Applications. Metabolites 2022, 12, 50. [Google Scholar] [CrossRef]
- Ma, Z.; Zuo, T.; Frey, N.; Rangrez, A.Y. A systematic framework for understanding the microbiome in human health and disease: From basic principles to clinical translation. Sig. Transduct. Target Ther. 2024, 9, 237. [Google Scholar] [CrossRef]
- Aggarwal, N.; Kitano, S.; Puah, G.R.Y.; Kittelmann, S.; Hwang, I.Y.; Chang, M.W. Microbiome and Human Health: Current Understanding, Engineering, and Enabling Technologies. Chem. Rev. 2023, 123, 31–72. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Sig. Transduct. Target Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Marino, M.; Pellegrini, M.; La Rosa, P.; Acconcia, F. Susceptibility of Estrogen Receptor Rapid Responses to Xenoestrogens: Physiological Outcomes. Steroids 2012, 77, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, B.; Yang, J.; Zhou, J.; Xu, Y. Linear discriminant analysis. Nat. Rev. Methods Primers 2024, 4, 70. [Google Scholar] [CrossRef]
- Zeng, J.; Guo, Y.; Han, Y.; Li, Z.; Yang, Z.; Chai, Q.; Wang, W.; Zhang, Y.; Fu, C. A Review of the Discriminant Analysis Methods for Food Quality Based on Near-Infrared Spectroscopy and Pattern Recognition. Molecules 2021, 26, 749. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Balaban, A.T. Highly discriminating distance-based topological index. Chem. Phys. Lett. 1982, 89, 399–404. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: New York, NY, USA, 2006. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Radzi, S.F.M.; Karim, M.K.A.; Saripan, M.I.; Rahman, M.A.A.; Isa, I.N.C.; Ibahim, M.J. Hyperparameter Tuning and Pipeline Optimization via Grid Search Method and Tree-Based AutoML in Breast Cancer Prediction. J. Pers. Med. 2021, 11, 978. [Google Scholar] [CrossRef]
- McLachlan, G.J. Discriminant Analysis and Statistical Pattern Recognition; Wiley-Interscience: Hoboken, NJ, USA, 2004. [Google Scholar] [CrossRef]
- Bergstra, J.; Bengio, Y. Random Search for Hyper-Parameter Optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- He, H.; Garcia, E.A. Learning from Imbalanced Data. IEEE Trans. Knowl. Data Eng. 2009, 21, 1263–1284. [Google Scholar]
- Kuhn, M.; Johnson, K. Applied Predictive Modeling; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Cohen, J. A Coefficient of Agreement for Nominal Scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Zhang, Y.; Chang, W.; Zhou, M. The Effects and Mechanisms of Flavonoids on Cancer Prevention and Therapy: Focus on Gut Microbiota. Int. J. Biol. Sci. 2022, 18, 1451–1475. [Google Scholar] [CrossRef]
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef]
- Brylinski, M. Aromatic interactions at the ligand–protein interface: Implications for the development of docking scoring functions. Chem. Biol. Drug Des. 2018, 91, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Kier, L.B.; Hall, L.H. Molecular Connectivity in Structure-Activity Analysis; Research Studies Press: Letchworth, UK, 1986. [Google Scholar]
- Labute, P. A widely applicable set of descriptors. J. Mol. Graph. Model. 2000, 18, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Wilson, I.D. Opinion: Understanding ‘global’ systems biology: Metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003, 2, 668–676. [Google Scholar] [CrossRef]
- Steinkraus, K.H. Handbook of Indigenous Fermented Foods; Marcel Dekker Inc.: New York, NY, USA, 1996; ISBN 9780824793524. [Google Scholar]
- Negi, A.; Kesari, K.K.; Voisin-Chiret, A.S. Estrogen Receptor-α Targeting: PROTACs, SNIPERs, Peptide-PROTACs, Antibody Conjugated PROTACs and SNIPERs. Pharmaceutics 2022, 14, 2523. [Google Scholar] [CrossRef]
- Spaepen, S.; Vanderleyden, J.; Remans, R. Indole-3-acetic acid in microbial and microorganism–plant signaling. FEMS Microbiol. Rev. 2007, 31, 425–448. [Google Scholar] [CrossRef]
- Parr, A.J.; Bolwell, G.P. Phenols in the plant and in man. The potential for possible nutritional enhancement of the diet by modifying the phenols content or profile. J. Sci. Food Agric. 2000, 80, 985–1012. [Google Scholar] [CrossRef]
- Piwowarski, J.P.; Stanisławska, I.; Granica, S.; Stefańska, J.; Kiss, A.K. Phase II conjugates of urolithins isolated from human urine and potential role of β-glucuronidases in their disposition. Drug Metab. Dispos. 2017, 45, 657–665. [Google Scholar] [CrossRef]
- Selma, M.V.; Espín, J.C.; Tomás-Barberán, F.A. Interaction between phenolics and gut microbiota: Role in human health. J. Agric. Food Chem. 2009, 57, 6485–6501. [Google Scholar] [CrossRef]
- Zhang, A.Q.; Mitchell, S.C.; Smith, R.L. Dietary precursors of trimethylamine in man: A pilot study. Food Chem. Toxicol. 1999, 37, 515–520. [Google Scholar] [CrossRef]
- Toldrá, F. The role of muscle enzymes in dry-cured meat products with different drying conditions. Trends Food Sci. Technol. 2006, 17, 164–168. [Google Scholar] [CrossRef]
- Jay, J.M.; Loessner, M.J.; Golden, D.A. Modern Food Microbiology; Springer Science & Business Media: New York, NY, USA, 2005; ISBN 9780387231808. [Google Scholar]
- Davidson, P.M.; Sofos, J.N.; Branen, A.L. Antimicrobials in Food; CRC Press: Boca Raton, FL, USA, 2005; ISBN 9780824753337. [Google Scholar]
- Leroy, F.; De Vuyst, L. Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci. Technol. 2004, 15, 67–78. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.K.; Panda, S.H.; Singh, A.; Meena, D.K. Polyphenols in human health and disease: Current knowledge and future directions. J. Pharmacogn. Phytochem. 2015, 4, 17–27. [Google Scholar]
- Slavin, J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients 2013, 5, 1417–1435. [Google Scholar] [CrossRef]
- Adams, M.R.; Hall, C.J. Growth inhibition of food-borne pathogens by lactic and acetic acids and their mixtures. Int. J. Food Sci. Technol. 1988, 23, 287–292. [Google Scholar] [CrossRef]
| Model | Accuracy | Precision | Recall | F1 Score | MCC | Cohen’s Kappa | Hamming Loss | ROC AUC |
|---|---|---|---|---|---|---|---|---|
| LinearDiscriminantAnalysis | 0.8944 | 0.9000 | 0.9612 | 0.9296 | 0.7255 | 0.7192 | 0.1056 | 0.8396 |
| RidgeClassifier | 0.8873 | 0.8991 | 0.9515 | 0.9245 | 0.7074 | 0.7030 | 0.1127 | 0.8347 |
| LogisticRegression | 0.8662 | 0.8500 | 0.9903 | 0.9148 | 0.6522 | 0.6116 | 0.1338 | 0.7644 |
| Bagging | 0.8451 | 0.8716 | 0.9223 | 0.8962 | 0.5953 | 0.5916 | 0.1549 | 0.7817 |
| Bagging (DecisionTree) | 0.8451 | 0.8716 | 0.9223 | 0.8962 | 0.5953 | 0.5916 | 0.1549 | 0.7817 |
| BalancedRandomForest | 0.8239 | 0.9535 | 0.7961 | 0.8677 | 0.6334 | 0.6108 | 0.1761 | 0.8468 |
| CalibratedClassifierCV (Logistic) | 0.8239 | 0.8250 | 0.9612 | 0.8879 | 0.5214 | 0.4889 | 0.1761 | 0.7114 |
| AdaBoost | 0.8169 | 0.8235 | 0.9515 | 0.8829 | 0.5003 | 0.4733 | 0.1831 | 0.7065 |
| GradientBoosting | 0.8169 | 0.8468 | 0.9126 | 0.8785 | 0.5151 | 0.5092 | 0.1831 | 0.7384 |
| CatBoost | 0.8169 | 0.8532 | 0.9029 | 0.8774 | 0.5206 | 0.5174 | 0.1831 | 0.7463 |
| XGBoost | 0.8099 | 0.8455 | 0.9029 | 0.8732 | 0.4989 | 0.4946 | 0.1901 | 0.7335 |
| RandomForest | 0.8028 | 0.8319 | 0.9126 | 0.8704 | 0.4710 | 0.4623 | 0.1972 | 0.7127 |
| QuadraticDiscriminantAnalysis | 0.8028 | 0.8866 | 0.8350 | 0.8600 | 0.5304 | 0.5277 | 0.1972 | 0.7765 |
| EasyEnsemble | 0.7817 | 0.9286 | 0.7573 | 0.8342 | 0.5479 | 0.5241 | 0.2183 | 0.8017 |
| Voting | 0.7817 | 0.7951 | 0.9417 | 0.8622 | 0.3859 | 0.3544 | 0.2183 | 0.6504 |
| ExtraTree | 0.7676 | 0.8070 | 0.8932 | 0.8479 | 0.3692 | 0.3607 | 0.2324 | 0.6646 |
| KNeighbors | 0.7606 | 0.8000 | 0.8932 | 0.8440 | 0.3452 | 0.3355 | 0.2394 | 0.6517 |
| Bagging (KNeighbors) | 0.7465 | 0.7680 | 0.9320 | 0.8421 | 0.2591 | 0.2285 | 0.2535 | 0.5942 |
| DecisionTree | 0.7324 | 0.7982 | 0.8447 | 0.8208 | 0.2965 | 0.2946 | 0.2676 | 0.6403 |
| SGDClassifier | 0.7254 | 0.7254 | 1.0000 | 0.8408 | 0.0000 | 0.0000 | 0.2746 | 0.5000 |
| LinearSVC | 0.7254 | 0.7254 | 1.0000 | 0.8408 | 0.0000 | 0.0000 | 0.2746 | 0.5000 |
| SVC | 0.7254 | 0.7254 | 1.0000 | 0.8408 | 0.0000 | 0.0000 | 0.2746 | 0.5000 |
| MLPClassifier | 0.7254 | 0.7254 | 1.0000 | 0.8408 | 0.0000 | 0.0000 | 0.2746 | 0.5000 |
| CalibratedClassifierCV (SVC) | 0.7254 | 0.7254 | 1.0000 | 0.8408 | 0.0000 | 0.0000 | 0.2746 | 0.5000 |
| BernoulliNB | 0.7183 | 0.7603 | 0.8932 | 0.8214 | 0.1881 | 0.1747 | 0.2817 | 0.5748 |
| GaussianNB | 0.6056 | 1.0000 | 0.4563 | 0.6267 | 0.4328 | 0.3155 | 0.3944 | 0.7282 |
| GaussianProcess | 0.3732 | 0.8500 | 0.1650 | 0.2764 | 0.1131 | 0.0530 | 0.6268 | 0.5441 |
| No. | Feature | Importance |
|---|---|---|
| 1 | FractionCSP3 | 2.126618 |
| 2 | AromaticProportion | 2.126618 |
| 3 | BalabanJ | 0.895716 |
| 4 | NumHAcceptors | 0.698623 |
| 5 | Chi3v | 0.453634 |
| 6 | SMR_VSA2 | 0.431287 |
| 7 | Chi4v | 0.351189 |
| 8 | PEOE_VSA3 | 0.313827 |
| 9 | PEOE_VSA4 | 0.287226 |
| 10 | NOCount | 0.272061 |
| Metabolite | Predicted Activity |
|---|---|
| Indole | Active |
| Skatole (3-methylindole) | Active |
| Trimethylamine (TMA) | Active |
| Indole-3-acetic acid (IAA) | Active |
| Indole-3-lactate | Inactive |
| 4-Cresol (p-cresol) | Active |
| p-Cresol sulfate | Inactive |
| Phenylacetic acid | Inactive |
| Phenyllactic acid | Active |
| o-Cresol | Active |
| 3-Methylphenol (m-cresol) | Active |
| Deoxycholic acid | Inactive |
| Lithocholic acid | Inactive |
| Urolithin A | Active |
| Urolithin B | Active |
| Acetoin | Inactive |
| 2,3-Butanediol | Inactive |
| Cadaverine | Inactive |
| Putrescine | Inactive |
| Trimethylamine N-oxide (TMAO) | Active |
| Phenol | Active |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsov, M.; Chernyavskaya, O.; Kutuzov, M.; Vilkova, D.; Novichenko, O.; Stolyarova, A.; Mashin, D.; Nikitin, I. Development of a Predictive Model for the Biological Activity of Food and Microbial Metabolites Toward Estrogen Receptor Alpha (ERα) Using Machine Learning. Big Data Cogn. Comput. 2025, 9, 86. https://doi.org/10.3390/bdcc9040086
Kuznetsov M, Chernyavskaya O, Kutuzov M, Vilkova D, Novichenko O, Stolyarova A, Mashin D, Nikitin I. Development of a Predictive Model for the Biological Activity of Food and Microbial Metabolites Toward Estrogen Receptor Alpha (ERα) Using Machine Learning. Big Data and Cognitive Computing. 2025; 9(4):86. https://doi.org/10.3390/bdcc9040086
Chicago/Turabian StyleKuznetsov, Maksim, Olga Chernyavskaya, Mikhail Kutuzov, Daria Vilkova, Olga Novichenko, Alla Stolyarova, Dmitry Mashin, and Igor Nikitin. 2025. "Development of a Predictive Model for the Biological Activity of Food and Microbial Metabolites Toward Estrogen Receptor Alpha (ERα) Using Machine Learning" Big Data and Cognitive Computing 9, no. 4: 86. https://doi.org/10.3390/bdcc9040086
APA StyleKuznetsov, M., Chernyavskaya, O., Kutuzov, M., Vilkova, D., Novichenko, O., Stolyarova, A., Mashin, D., & Nikitin, I. (2025). Development of a Predictive Model for the Biological Activity of Food and Microbial Metabolites Toward Estrogen Receptor Alpha (ERα) Using Machine Learning. Big Data and Cognitive Computing, 9(4), 86. https://doi.org/10.3390/bdcc9040086