
Exploring Evolutionary Relationships within Neodermata Using Putative Orthologous Groups of Proteins, with Emphasis on Peptidases

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Evolutionary Relationships of Neodermata
2.1.1. Phylogenetic Analysis of BUSCO and OMA OGPs
2.1.2. Phylogenetic Analysis of Peptidases
2.1.3. Network Analysis of Peptidase OGs
2.1.4. Hierarchical Grouping Analysis in Principal Components of OGs
2.2. Mutation Rates in Secreted and Non-Secreted Peptidases
2.3. Classification of the C01A and S01C Peptidase Subfamilies
3. Results
3.1. Evolutionary Relationships of Neodermata
3.1.1. Phylogenetic Analyses of BUSCO and OMA OGPs
3.1.2. Phylogenetic Analysis of Peptidases
3.1.3. Network Analysis and Hierarchical Grouping Analysis in Principal Components of Orthologous Groups
3.2. Mutation Rate in Secreted and Non-Secreted Peptidases
3.2.1. Single-Copy Orthologous Groups vs. Multiple-Copy Orthologous Groups
3.2.2. Single-Copy Secreted Protein Orthologous Groups vs. Single-Copy Non-Secreted Proteins Orthologous Groups
3.2.3. Multiple-Copy Orthologous Groups Secreted Proteins vs. Multiple-Copy Orthologous Groups Non-Secreted Proteins
3.3. Classification of the C01A Peptidase Subfamily and S2 Active Subsite Residues
3.4. Classification of the S01C Peptidase Subfamily
4. Discussion
4.1. Non-Monophyly of Monogenea
4.2. Monopisthocotylea + Cestoda
4.3. Polyopisthocotylea + Trematoda
4.4. Peptidases in Neodermata
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hahn, C.; Fromm, B.; Bachmann, L. Comparative genomics of flatworms (Platyhelminthes) reveals shared genomic features of ecto-and endoparastic neodermata. Genome Biol. Evol. 2014, 6, 1105–1117. [Google Scholar] [CrossRef]
- Littlewood, D.T.J.; Rohde, K.; Clough, K.A. Interrelationships of all major groups of Platyhelminthes: Phylogenetic evidence from morphology and molecules. Biol. J. Linn. Soc. 1999, 66, 75–114. [Google Scholar] [CrossRef]
- Perkins, E.M.; Donnellan, S.C.; Bertozzi, T.; Whittington, I.D. Closing the mitochondrial circle on paraphyly of the Monogenea (Platyhelminthes) infers evolution in the diet of parasitic flatworms. Int. J. Parasitol. 2010, 40, 1237–1245. [Google Scholar] [CrossRef]
- Zhang, D.; Li, W.X.; Zou, H.; Wu, S.G.; Li, M.; Jakovlić, I.; Zhang, J.; Chen, R.; Wang, G. Homoplasy or plesiomorphy? Reconstruction of the evolutionary history of mitochondrial gene order rearrangements in the subphylum Neodermata. Int. J. Parasitol. 2019, 49, 819–829. [Google Scholar] [CrossRef]
- Boeger, W.A.; Kritsky, D.C. Phylogenetic relationships of the Monogenoidea. In Interrelationships of the Platyhelminthes; Littlewood, D.T.J., Bray, R.A., Eds.; Taylor and Francis: London, UK, 2001; pp. 92–102. [Google Scholar]
- Lockyer, A.E.; Olson, P.D.; Littlewood, D.T.J. Utility of complete large and small subunit rRNA genes in resolving the phylogeny of the Neodermata (Platyhelminthes): Implications and a review of the cercomer theory. Biol. J. Linn. Soc. 2003, 78, 155–171. [Google Scholar] [CrossRef]
- Boeger, W.A.; Kritsky, D.C. Phylogeny and a revised classification of the Monogenoidea Bychowsky 1937 (Platyhelminthes). Syst. Parasitol. 1993, 26, 1–32. [Google Scholar] [CrossRef]
- Justine, J.L. Cladistic study in the Monogenea “Platyhelminthes” based upon a parsimony analysis of spermiogenetic and spermatozoal ultrastructural characters. Int. J. Parasitol. 1991, 21, 821–838. [Google Scholar] [CrossRef]
- Justine, J.L. Non-monophyly of the monogeneans? Int. J. Parasitol. 1998, 28, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Laumer, C.E.; Giribet, G. Inclusive taxon sampling suggests a single, stepwise origin of ectolecithality in Platyhelminthes. Biol. J. Linn. Soc. 2014, 111, 570–588. [Google Scholar] [CrossRef]
- Park, J.K.; Kim, K.H.; Kang, S.; Kim, W.; Eom, K.S.; Littlewood, D.T.J. A common origin of complex life cycles in parasitic flatworms: Evidence from the complete mitochondrial genome of Microcotyle sebastis (Monogenea: Platyhelminthes). BMC Evol. Biol. 2007, 7, 1–13. [Google Scholar] [CrossRef]
- Laumer, C.E.; Hejnol, A.; Giribet, G. Nuclear genomic signals of the ‘microturbellarian’ roots of platyhelminth evolutionary innovation. eLife 2015, 4, e05503. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Bleidorn, C.; Braband, A.; Dambach, J.; Donath, A.; Fritzsch, G.; Golombek, A.; Hadrys, H.; Jühling, F.; Meusemann, K.; et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Biol. Evol. 2013, 69, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Charrier, N.P.; Hermouet, A.; Hervet, C.; Agoulon, A.; Barker, S.C.; Heylen, D.; Toty, C.; McCoy, K.D.; Plantard, O.; Rispe, C. A transcriptome-based phylogenetic study of hard ticks (Ixodidae). Sci. Rep. 2019, 9, 12923. [Google Scholar] [CrossRef]
- Hawkins, J.A.; Kaczmarek, M.E.; Müller, M.A.; Drosten, C.; Press, W.H.; Sawyer, S.L. A metaanalysis of bat phylogenetics and positive selection based on genomes and transcriptomes from 18 species. Proc. Natl Acad. Sci. USA 2019, 116, 11351–11360. [Google Scholar] [CrossRef] [PubMed]
- Gadagkar, S.R.; Rosenberg, M.S.; Kumar, S. Inferring species phylogenies from multiple genes: Concatenated sequence tree versus consensus gene tree. J. Exp. Zool. B Mol. Dev. Evol. 2005, 304, 64–74. [Google Scholar] [CrossRef]
- Philippe, H.; de Vienne, D.M.; Ranwez, V.; Roure, B.; Baurain, D.; Delsuc, F. Pitfalls in supermatrix phylogenomics. Eur. J. Taxon. 2017, 283, 1–25. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Dylus, D.; Nevers, Y.; Altenhoff, A.M.; Gürtler, A.; Dessimoz, C.; Glover, N.M. How to build phylogenetic species trees with OMA. F1000Research 2020, 9, 511. [Google Scholar] [CrossRef]
- Patwardhan, A.; Ray, S.; Roy, A. Molecular markers in phylogenetic studies-a review. J. Phylogen. Evol. Biol. 2014, 2, 131. [Google Scholar] [CrossRef]
- Kasný, M.; Mikes, L.; Hampl, V.; Dvorak, J.; Caffrey, C.R.; Dalton, J.P.; Horak, P. Peptidases of trematodes. Adv. Parasitol. 2009, 69, 205–297. [Google Scholar] [CrossRef]
- Dvorak, J.; Horn, M. Serine proteases in schistosomes and other trematodes. Int. J. Parasitol. 2018, 48, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Jedličková, L.; Dvořáková, H.; Dvořák, J.; Kašný, M.; Ulrychová, L.; Vorel, J.; Žárský, V.; Mikeš, L. Cysteine peptidases of Eudiplozoon nipponicum: A broad repertoire of structurally assorted cathepsins L in contrast to the scarcity of cathepsins B in an invasive species of haematophagous monogenean of common carp. Parasit. Vectors 2018, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, S.L.; McManus, D.P. Protease inhibitors of parasitic flukes: Emerging roles in parasite survival and immune defence. Trends Parasitol. 2017, 33, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, C.R.; Goupil, L.; Rebello, K.M.; Dalton, J.P.; Smith, D. Cysteine proteases as digestive enzymes in parasitic helminths. PLoS Negl. Trop. Dis. 2018, 12, e0005840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Robinson, M.W.; Dalton, J.P.; Donnelly, S. Helminth pathogen cathepsin proteases: It’s a family affair. Trends Biochem. Sci. 2008, 33, 601–608. [Google Scholar] [CrossRef]
- Caña-Bozada, V.; Morales-Serna, F.N.; Fajer-Ávila, E.J.; Llera-Herrera, R. De novo transcriptome assembly and identification of GPCRs in two species of monogenean parasites of fish. Parasite 2022, 29, 51. [Google Scholar] [CrossRef]
- Howe, K.L.; Bolt, B.J.; Shafie, M.; Kersey, P.; Berriman, M. WormBase ParaSite—A comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 2017, 215, 2–10. [Google Scholar] [CrossRef]
- Vorel, J.; Cwiklinski, K.; Roudnický, P.; Ilgová, J.; Jedličková, L.; Dalton, J.P.; Mikeš, L.; Gelnar, M.; Kašný, M. Eudiplozoon nipponicum (Monogenea, Diplozoidae) and its adaptation to haematophagy as revealed by transcriptome and secretome profiling. BMC Genom. 2021, 22, 274. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Levy, J.; Zarowiecki, M.; Tomiczek, B.; Vesztrocy, A.W.; Dalquen, D.A.; Müller, S.; Telford, M.J.; Glover, N.M.; Dylus, D.; et al. OMA standalone: Orthology inference among public and custom genomes and transcriptomes. Genome Res. 2019, 29, 1152–1163. [Google Scholar] [CrossRef]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Campos, A.; Cummings, M.P.; Reyes, J.L.; Laclette, J.P. Phylogenetic relationships of Platyhelminthes based on 18S ribosomal gene sequences. Mol. Biol. Evol. 1998, 10, 1–10. [Google Scholar] [CrossRef]
- Kishino, H.; Miyata, T.; Hasegawa, M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol. 1990, 31, 151–160. [Google Scholar] [CrossRef]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Strimmer, K.; Rambaut, A. Inferring confidence sets of possibly misspecified gene trees. Proc. Biol. Sci. 2002, 269, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, Z.; Hu, Y. The repertoire of G-protein-coupled receptors in Xenopus tropicalis. BMC Genom. 2009, 10, 263. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; Von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004, 17, 349–356. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, Atlanta, GA, USA, 6–9 June 2009; Volume 3, pp. 361–362. [Google Scholar]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Fortunato, S. Community detection in graphs. Phys. Rep. 2010, 486, 75–174. [Google Scholar] [CrossRef]
- Lázár, A.; Abel, D.; Vicsek, T. Modularity measure of networks with overlapping communities. Europhys. Lett. 2010, 90, 18001. [Google Scholar] [CrossRef]
- Husson, F.; Josse, J.; Le, S.; Mazet, J.; Husson, M.F. Package ‘FactoMineR’. R Package 2016, 96, 698. [Google Scholar]
- Odong, T.L.; Van Heerwaarden, J.; van Hintum, T.J.L.; van Eeuwijk, F.A.; Jansen, J. Improving hierarchical clustering of genotypic data via principal component analysis. Crop Sci. 2013, 53, 1546–1554. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Caña-Bozada, V.; Morales-Serna, F.N. PANAS: Pipeline and a case study to obtain synonymous and nonsynonymous substitution rates in genes of Platyhelminthes. Comp. Parasitol. 2023; in press. [Google Scholar]
- Wang, S.; Wang, S.; Luo, Y.; Xiao, L.; Luo, X.; Gao, S.; Dou, Y.; Zhang, H.; Guo, A.; Meng, Q.; et al. Comparative genomics reveals adaptive evolution of Asian tapeworm in switching to a new intermediate host. Nat. Commun. 2016, 7, 12845. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Stack, C.M.; Caffrey, C.R.; Donnelly, S.M.; Seshaadri, A.; Lowther, J.; Tort, J.F.; Collins, P.R.; Robinson, M.W.; Xu, W.; McKerrow, J.H.; et al. Structural and functional relationships in the virulence-associated cathepsin L proteases of the parasitic liver fluke, Fasciola hepatica. J. Biol. Chem. 2008, 283, 9896–9908. [Google Scholar] [CrossRef]
- Ingram, J.R.; Rafi, S.B.; Eroy-Reveles, A.A.; Ray, M.; Lambeth, L.; Hsieh, I.; Ruelas, D.; Lim, K.C.; Sakanari, J.; Craik, C.S.; et al. Investigation of the proteolytic functions of an expanded cercarial elastase gene family in Schistosoma mansoni. PLoS Negl. Trop. Dis. 2012, 6, e1589. [Google Scholar] [CrossRef]
- Cwiklinski, K.; Donnelly, S.; Drysdale, O.; Jewhurst, H.; Smith, D.; Verissimo, C.D.M.; Pritsch, I.C.; O’Neill, S.; Dalton, J.P.; Robinson, M.W. The cathepsin-like cysteine peptidases of trematodes of the genus Fasciola. Adv. Parasitol. 2019, 104, 113–164. [Google Scholar] [CrossRef]
- Turk, D.; Gunčar, G.; Podobnik, M.; Turk, B. Revised definition of substrate binding sites of papain-like cysteine proteases. Biol. Chem. 1998, 379, 137–147. [Google Scholar] [CrossRef]
- Lecaille, F.; Choe, Y.; Brandt, W.; Li, Z.; Craik, C.S.; Brömme, D. Selective inhibition of the collagenolytic activity of human cathepsin K by altering its S2 subsite specificity. Biochemistry 2002, 41, 8447–8454. [Google Scholar] [CrossRef]
- Corvo, I.; Cancela, M.; Cappetta, M.; Pi-Denis, N.; Tort, J.F.; Roche, L. The major cathepsin L secreted by the invasive juvenile Fasciola hepatica prefers proline in the S2 subsite and can cleave collagen. Mol. Biochem. Parasitol. 2009, 167, 41–47. [Google Scholar] [CrossRef]
- Robinson, M.W.; Corvo, I.; Jones, P.M.; George, A.M.; Padula, M.P.; To, J.; Cancela, M.; Rinaldi, G.; Tort, J.F.; Roche, L.; et al. Collagenolytic activities of the major secreted cathepsin L peptidases involved in the virulence of the helminth pathogen, Fasciola hepatica. PLoS Negl. Trop. Dis. 2011, 5, e1012. [Google Scholar] [CrossRef]
- Lambert, A. Oncomiracidiums et phylogenèse des Monogenea (Plathelminthes)-lre Partie: Développement post-larvaire. Ann. Parasitol. Hum. Comp. 1980, 55, 165–198. [Google Scholar] [CrossRef]
- Brooks, D.R.; O’Grady, R.T.; Glen, D.R. The phylogeny of the cercomeria Brooks, 1982 (Platyhelminthes). Proc. Helminthol. Soc. Wash. 1985, 52, 606–616. [Google Scholar]
- Ehlers, U. Comments on a phylogenetic system of the Platyhelminthes. In Advances in the Biology of Turbellarians and Related Platyhelminthes. Developments in Hydrobiology; Tyler, S., Ed.; Springer: Berlin/Heidelberg, Germany, 1986; Volume 32. [Google Scholar] [CrossRef]
- Rohde, K. Phylogeny of Platyhelminthes, with special reference to parasitic groups. Int. J. Parasitol. 1990, 20, 979–1007. [Google Scholar] [CrossRef]
- Justine, J.L.; Lambert, A.; Mattei, X. Spermatozoon ultrastructure and phylogenetic relationships in the monogeneans (Platyhelminthes). Int. J. Parasitol. 1985, 15, 601–608. [Google Scholar] [CrossRef]
- Baverstock, P.R.; Fielke, R.; Johnson, A.M.; Bray, R.A. Beveridge I. Conflicting phylogenetic hypotheses for the parasitic platyhelminths tested by partial sequencing of 18S ribosomal RNA. Int. J. Parasitol. 1991, 21, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Rohde, K.; Hefford, C.; Ellis, J.T.; Baverstock, P.R.; Johnson, A.M.; Watson, N.A.; Dittmann, S. Contributions to the phylogeny of Platyhelminthes based on partial sequencing of 18S ribosomal DNA. Int. J. Parasitol. 1993, 23, 705–724. [Google Scholar] [CrossRef]
- Mollaret, I.; Jamieson, B.G.; Adlard, R.D.; Hugall, A.; Lecointre, G.; Chombard, C.; Justine, J.L. Phylogenetic analysis of the Monogenea and their relationships with Digenea and Eucestoda inferred from 28S rDNA sequences. Mol. Biochem. Parasitol. 1997, 90, 433–438. [Google Scholar] [CrossRef]
- Litvaitis, M.K.; Rohde, K. A molecular test of platyhelminth phylogeny: Inferences from partial 28s rDNA sequences. Invertebr. Biol. 1999, 118, 42–56. [Google Scholar] [CrossRef]
- International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef]
- Littlewood, D.T.J.; Rohde, K.; Bray, R.A.; Herniou, E.A. Phylogeny of the Platyhelminthes and the evolution of parasitism. Biol. J. Linn. Soc. 1999, 68, 257–287. [Google Scholar] [CrossRef]
- Bychowsky, B.E. Ontogenesis and phylogenetic interrelations of parasitic flatworms. Izvest. Akad. Nauk SSSR Ser. Biol. 1937, 4, 1353–1383. [Google Scholar]
- Xylander, W.E.R. The Gyrocotylidea, Amphilinidea and the early evolution of Cestoda. In Interrelationships of the Platyhelminthes; Littlewood, D.T.J., Bray, R.A., Eds.; Taylor and Francis, Inc.: New York, NY, USA, 2001; pp. 103–111. [Google Scholar]
- Jedličková, L.; Dvořák, J.; Hrachovinová, I.; Ulrychová, L.; Kašný, M.; Mikeš, L. A novel Kunitz protein with proposed dual function from Eudiplozoon nipponicum (Monogenea) impairs haemostasis and action of complement in vitro. Int. J. Parasitol. 2019, 49, 337–346. [Google Scholar] [CrossRef]
- Dalton, J.P.; Skelly, P.; Halton, D.W. Role of the tegument and gut in nutrient uptake by parasitic platyhelminths. Can. J. Zool. 2004, 82, 211–232. [Google Scholar] [CrossRef]
- Hirazawa, N.; Umeda, N.; Hatanaka, A.; Kuroda, A. Characterization of serine proteases in the monogenean Neobenedenia girellae. Aquaculture 2006, 255, 188–195. [Google Scholar] [CrossRef]
- Rogozhin, E.A.; Solovyev, M.M.; Frolova, T.V.; Izvekova, G.I. Isolation and partial structural characterization of new Kunitz-type trypsin inhibitors from the pike cestode Triaenophorus nodulosus. Mol. Biochem. Parasitol. 2019, 233, 111217. [Google Scholar] [CrossRef] [PubMed]
- Izvekova, G.I.; Frolova, T.V.; Izvekov, E.I. Adsorption and inactivation of proteolytic enzymes by Triaenophorus nodulosus (Cestoda). Helminthologia 2017, 54, 3–10. [Google Scholar] [CrossRef]
- Li, Y.D.; Xie, Z.Y.; Du, Y.L.; Zhou, Z.; Mao, X.M.; Lv, L.X.; Li, Y.Q. The rapid evolution of signal peptides is mainly caused by relaxed selection on non-synonymous and synonymous sites. Gene 2009, 436, 8–11. [Google Scholar] [CrossRef]
- Jedličková, L.; Dvořáková, H.; Kašný, M.; Ilgová, J.; Potěšil, D.; Zdráhal, Z.; Mikeš, L. Major acid endopeptidases of the blood-feeding monogenean Eudiplozoon nipponicum (Heteronchoinea: Diplozoidae). Parasitology 2016, 143, 494–506. [Google Scholar] [CrossRef]
- Lowther, J.; Robinson, M.W.; Donnelly, S.M.; Xu, W.; Stack, C.M.; Matthews, J.M.; Dalton, J.P. The importance of pH in regulating the function of the Fasciola hepatica cathepsin L1 cysteine protease. PLoS Negl. Trop. Dis. 2009, 3, e369. [Google Scholar] [CrossRef]
- Kawatsu, H. Studies on the anemia of fish–IX. Hypochromic microcytic anemia of crucian carp caused by infestation with a trematode, Diplozoon nipponicum. Bull. Japan. Soc. Sci. Fish. 1978, 44, 1315–1319. [Google Scholar] [CrossRef]
- Thoney, D.A.; Hargis Jr, W.J. Monogenea (Platyhelminthes) as hazards for fish in confinement. Annu. Rev. Fish Dis. 1991, 1, 133–153. [Google Scholar] [CrossRef]
- Morales-Serna, F.N.; Martínez-Brown, J.M.; Avalos-Soriano, A.; Sarmiento-Vázquez, S.; Hernández-Inda, Z.L.; Medina-Guerrero, R.M.; Fajer-Ávila, E.J.; Ibarra-Castro, L. The efficacy of Geraniol and ß-Citronellol against freshwater and marine monogeneans. J. Aquati. Anim. Health 2020, 32, 127–132. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
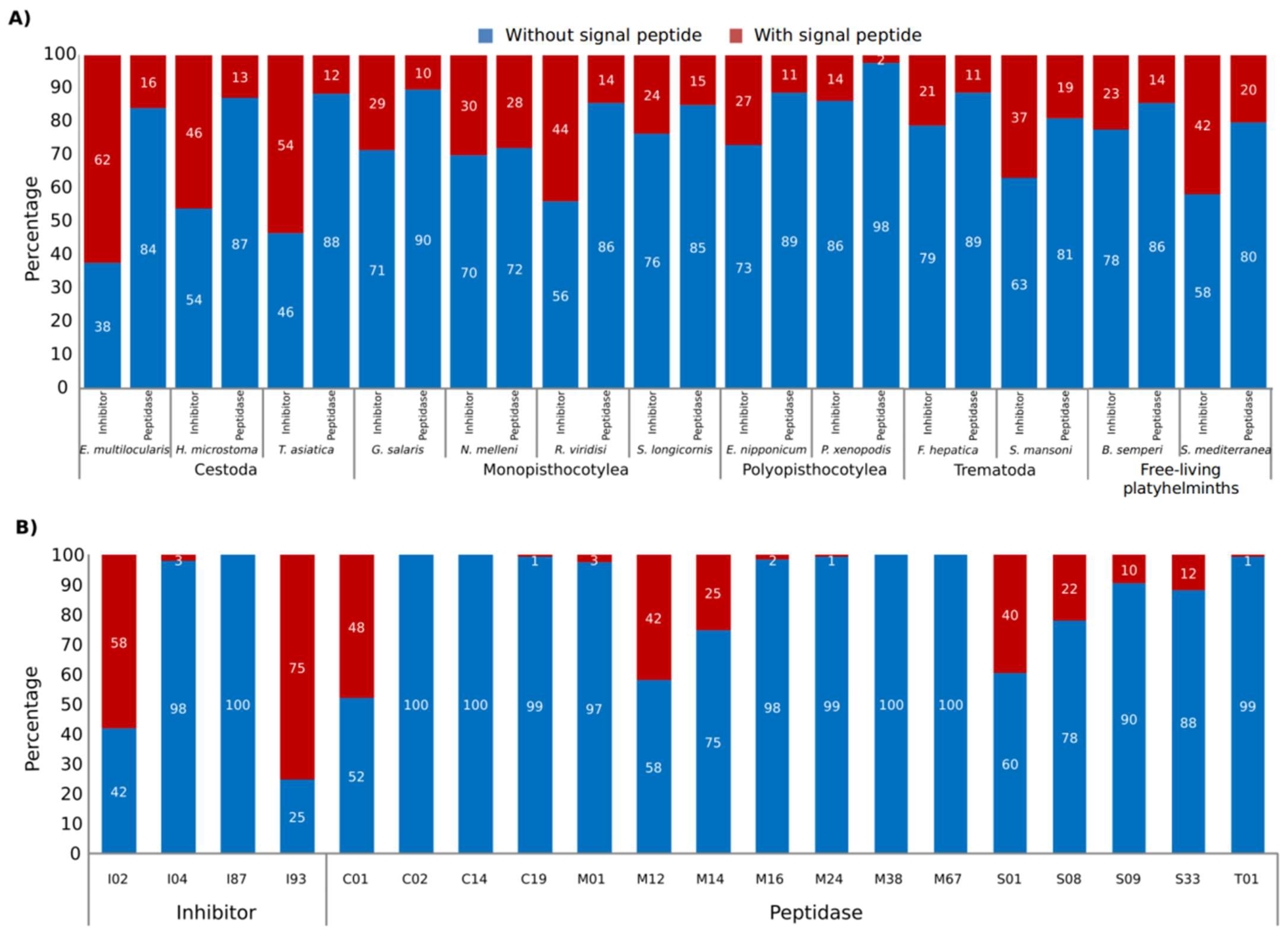{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenario | Constrained Tree | logL | deltaL | bp-RELL | p-KH | p-SH | c-ELW | p-AU | Reference |
|---|---|---|---|---|---|---|---|---|---|
| BUSCO OGPs | |||||||||
| 1 | ((Monopisthocotylea, Cestoda), (Polyopisthocotylea, Trematoda)) | −504,202.9113 | 0 | 0.941+ | 0.943+ | 1+ | 0.938+ | 0.947+ | Present study |
| 2 | ((Monopisthocotylea, Polyopisthocotylea), (Cestoda, Trematoda)) | −504,265.679 | 62.768 | 0.0137− | 0.0567+ | 0.0567+ | 0.0205− | 0.0542+ | [6] |
| 3 | (((Monopisthocotylea, Polyopisthocotylea), Cestoda), Trematoda) | −504,265.679 | 62.768 | 0.0208− | 0.0567+ | 0.0567+ | 0.0205− | 0.0538+ | [42] |
| 4 | (((Trematoda, Cestoda), Polyopisthocotylea), Monopisthocotylea) | −504,265.679 | 62.768 | 0.0248+ | 0.0567+ | 0.0567+ | 0.0205+ | 0.0534+ | [3] |
| OMA OGPs | |||||||||
| 1 | ((Monopisthocotylea, Cestoda), (Polyopisthocotylea, Trematoda)) | −2,015,132.409 | 0 | 0.987+ | 0.987+ | 1+ | 0.986+ | 0.986+ | Present study |
| 2 | ((Monopisthocotylea, Polyopisthocotylea), (Cestoda, Trematoda)) | −2,015,328.208 | 195.8 | 0.0029− | 0.0131− | 0.0131− | 0.00454− | 0.0142− | [6] |
| 3 | (((Monopisthocotylea, Polyopisthocotylea), Cestoda), Trematoda) | −2,015,328.208 | 195.8 | 0.0044− | 0.0131− | 0.0131− | 0.00454− | 0.0142− | [42] |
| 4 | (((Trematoda, Cestoda), Polyopisthocotylea), Monopisthocotylea) | −2,015,328.208 | 195.8 | 0.0062− | 0.0131− | 0.0131− | 0.00454− | 0.0144− | [3] |
| C01A Subfamily | S01C Subfamily | |||
|---|---|---|---|---|
| Species | Cathepsin B | Cathepsin C | Cathepsin L | Cercarial Elastase |
| E. multilocularis | 2 | 0 | 4 | 0 |
| H. microstoma | 2 | 0 | 4 | 0 |
| T. asiatica | 3 | 0 | 7 | 0 |
| G. salaris | 3 | 3 | 7 | 19 |
| N. melleni | 10 | 1 | 5 | 37 |
| R. viridisi | 2 | 1 | 3 | 8 |
| S. longicornis | 6 | 2 | 14 | 0 |
| E. nipponicum | 2 | 5 | 12 | 1 |
| P. xenopodis | 6 | 3 | 7 | 0 |
| F. hepatica | 17 | 0 | 16 | 1 |
| S. mansoni | 6 | 1 | 6 | 5 |
| B. semperi | 9 | 1 | 16 | 0 |
| S. mediterranea | 2 | 2 | 20 | 0 |
| Total | 70 | 19 | 121 | 71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caña-Bozada, V.; Robinson, M.W.; Hernández-Mena, D.I.; Morales-Serna, F.N. Exploring Evolutionary Relationships within Neodermata Using Putative Orthologous Groups of Proteins, with Emphasis on Peptidases. Trop. Med. Infect. Dis. 2023, 8, 59. https://doi.org/10.3390/tropicalmed8010059
Caña-Bozada V, Robinson MW, Hernández-Mena DI, Morales-Serna FN. Exploring Evolutionary Relationships within Neodermata Using Putative Orthologous Groups of Proteins, with Emphasis on Peptidases. Tropical Medicine and Infectious Disease. 2023; 8(1):59. https://doi.org/10.3390/tropicalmed8010059
Chicago/Turabian StyleCaña-Bozada, Víctor, Mark W. Robinson, David I. Hernández-Mena, and Francisco N. Morales-Serna. 2023. "Exploring Evolutionary Relationships within Neodermata Using Putative Orthologous Groups of Proteins, with Emphasis on Peptidases" Tropical Medicine and Infectious Disease 8, no. 1: 59. https://doi.org/10.3390/tropicalmed8010059
APA StyleCaña-Bozada, V., Robinson, M. W., Hernández-Mena, D. I., & Morales-Serna, F. N. (2023). Exploring Evolutionary Relationships within Neodermata Using Putative Orthologous Groups of Proteins, with Emphasis on Peptidases. Tropical Medicine and Infectious Disease, 8(1), 59. https://doi.org/10.3390/tropicalmed8010059