Anti-Trypanosomal Proteasome Inhibitors Cure Hemolymphatic and Meningoencephalic Murine Infection Models of African Trypanosomiasis

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Parasites, Cell Culture and Growth Inhibition Assays
2.2. HepG2 Cytotoxicity Assay
2.3. Determination of Solubility, PAMPA, Plasma Protein Binding, Brain Tissue Binding and Microsomal Clearance
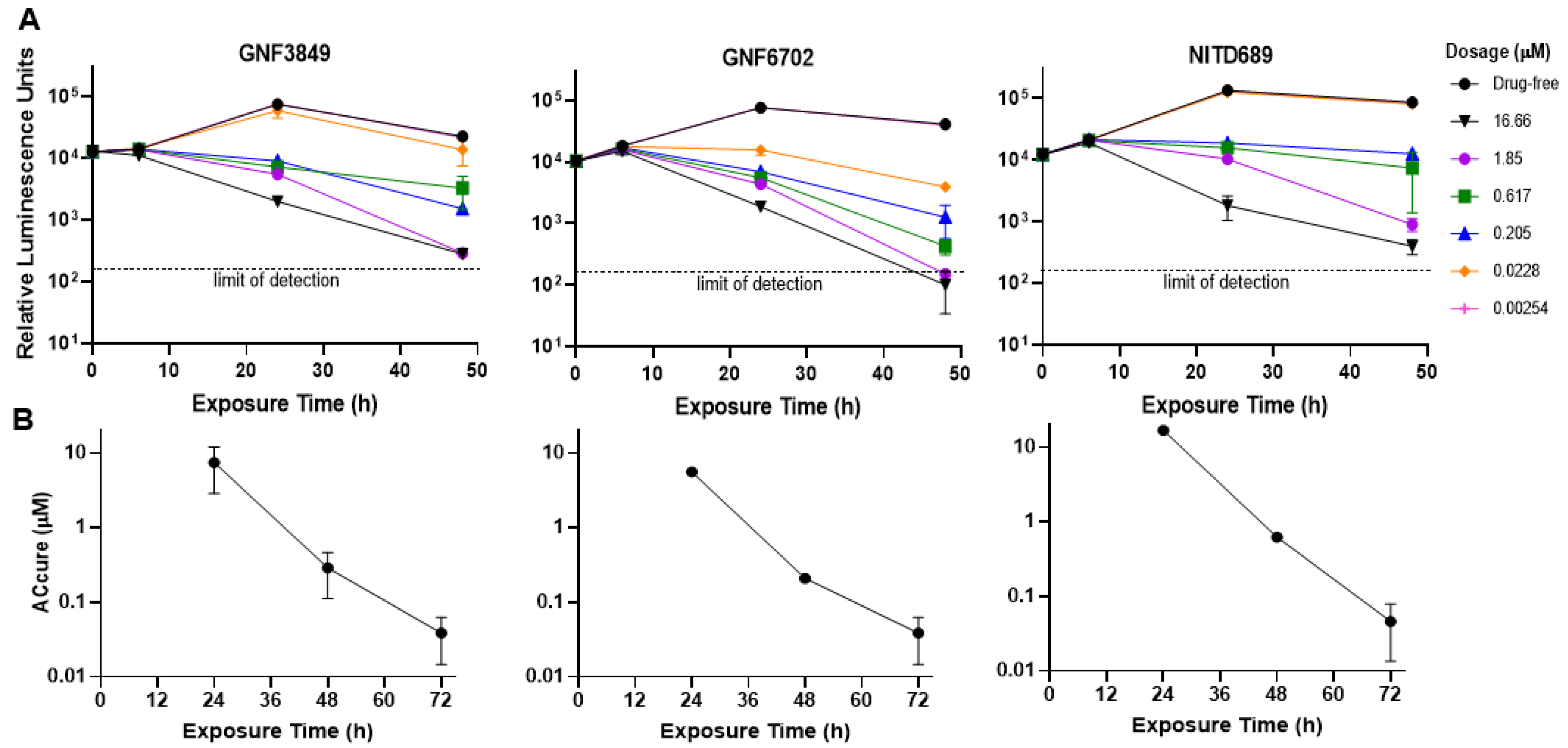
2.4. Time to Kill and Reversibility Assays
2.5. In Vivo Pharmacokinetic (PK) Analysis
2.6. Hemolymphatic Mouse Model (Stage 1 HAT Efficacy Model)
2.7. Meningocephalic Mouse Model (Stage 2 HAT Efficacy Model)
3. Results
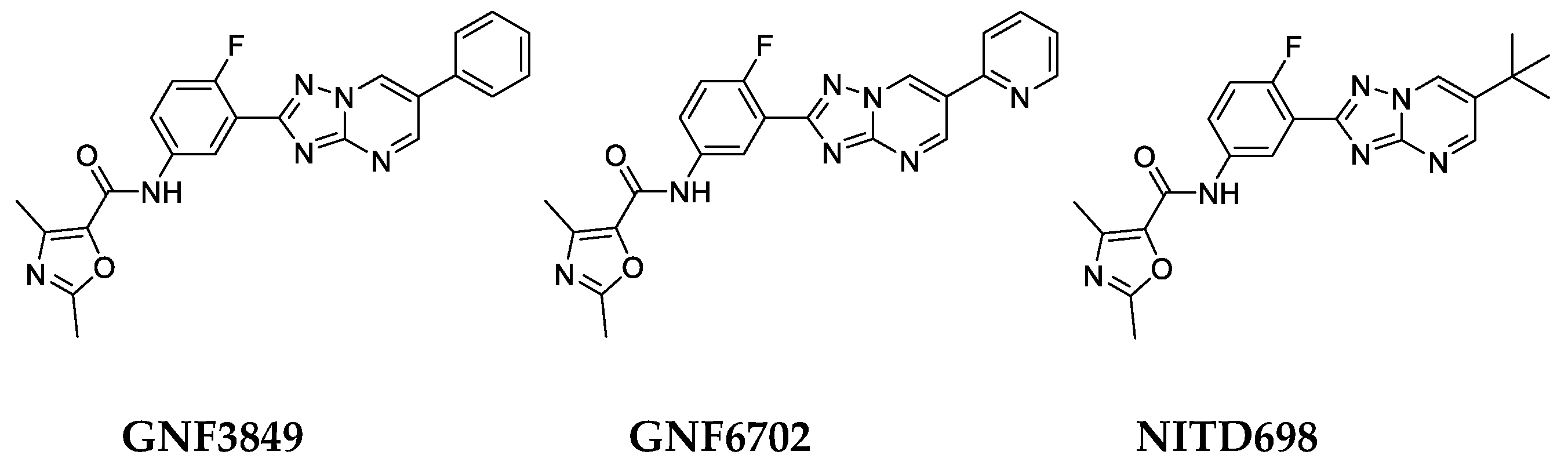
3.1. Extended Characterization of TP Series of Kinetoplastid Proteasome Inhibitors for Treatment of Human African Trypanosomiasis
3.2. Biological Profiling of the TP Class of Inhibitors
3.3. The TP Class of Inhibitors Are Active against Clinical Isolates
3.4. The TP Class of Compounds Are Proteasome Inhibitors
3.5. In Vivo Pharmacokinetic Properties of TP Class of Compounds
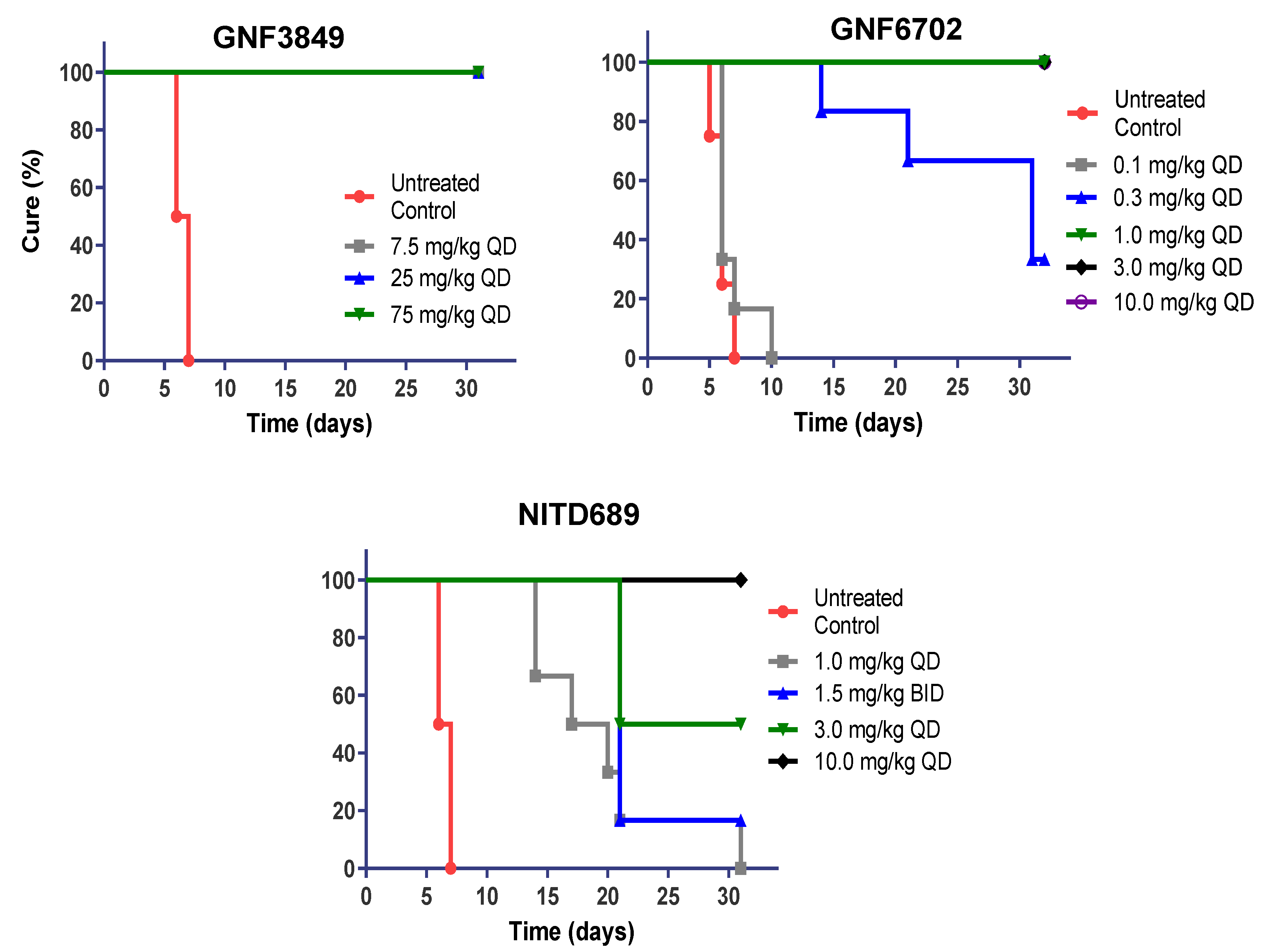
3.6. TP Class of Compounds Is Efficacious against Hemolymphatic Infection in a Stage 1 HAT Mouse Model
3.7. Assessment of Brain Permeability, Tissue Binding and Partitioning of TP Compounds
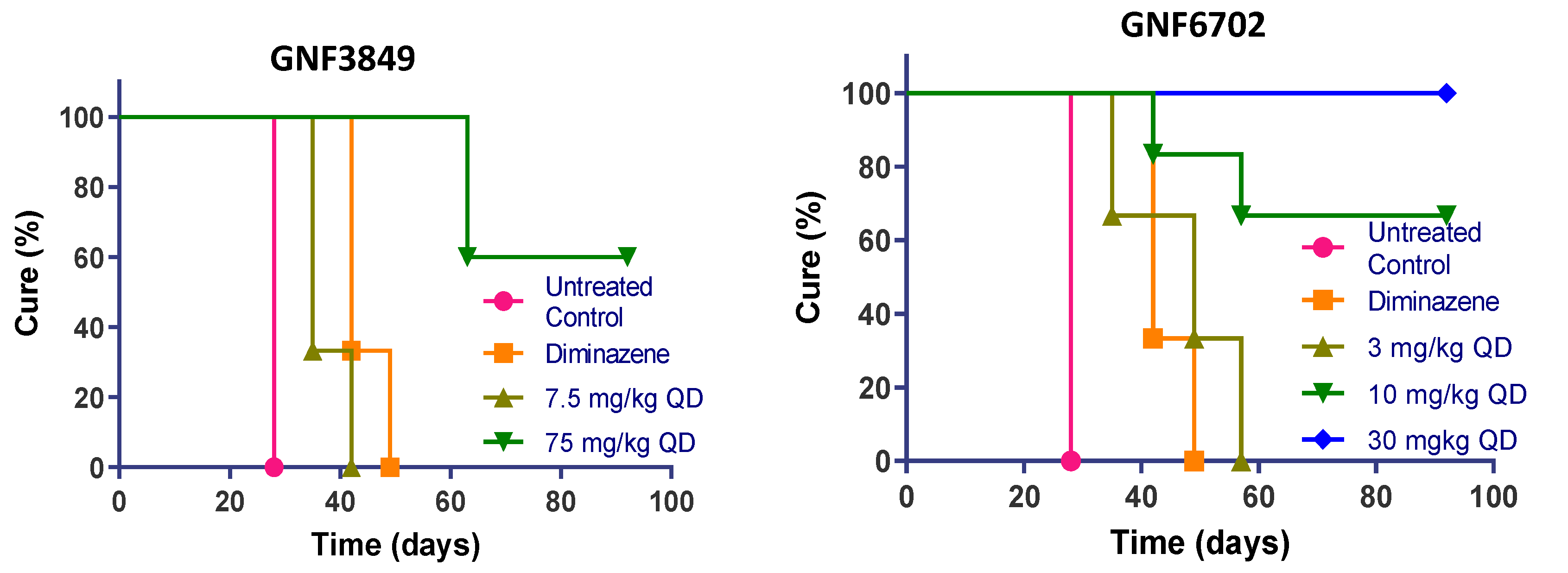
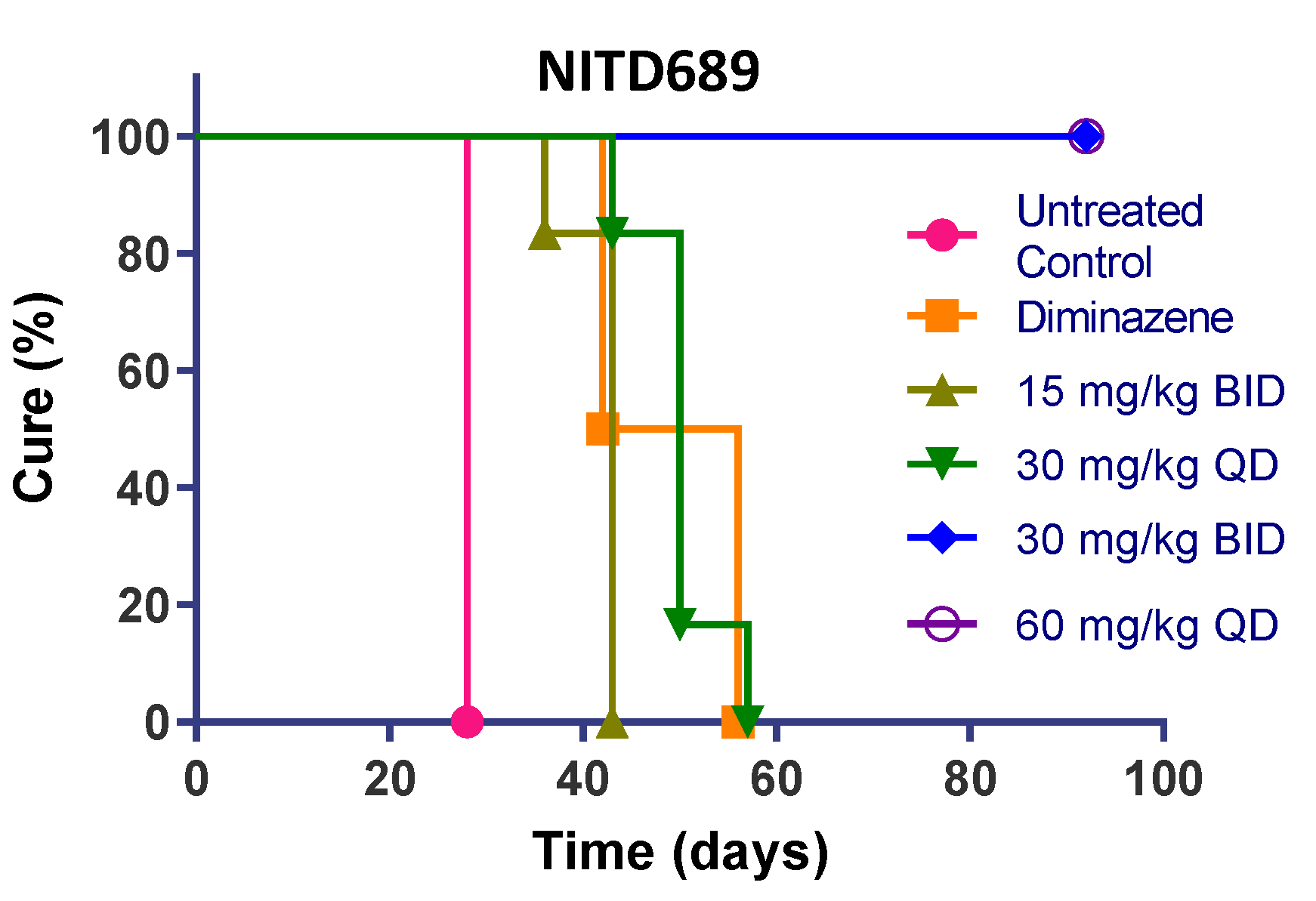
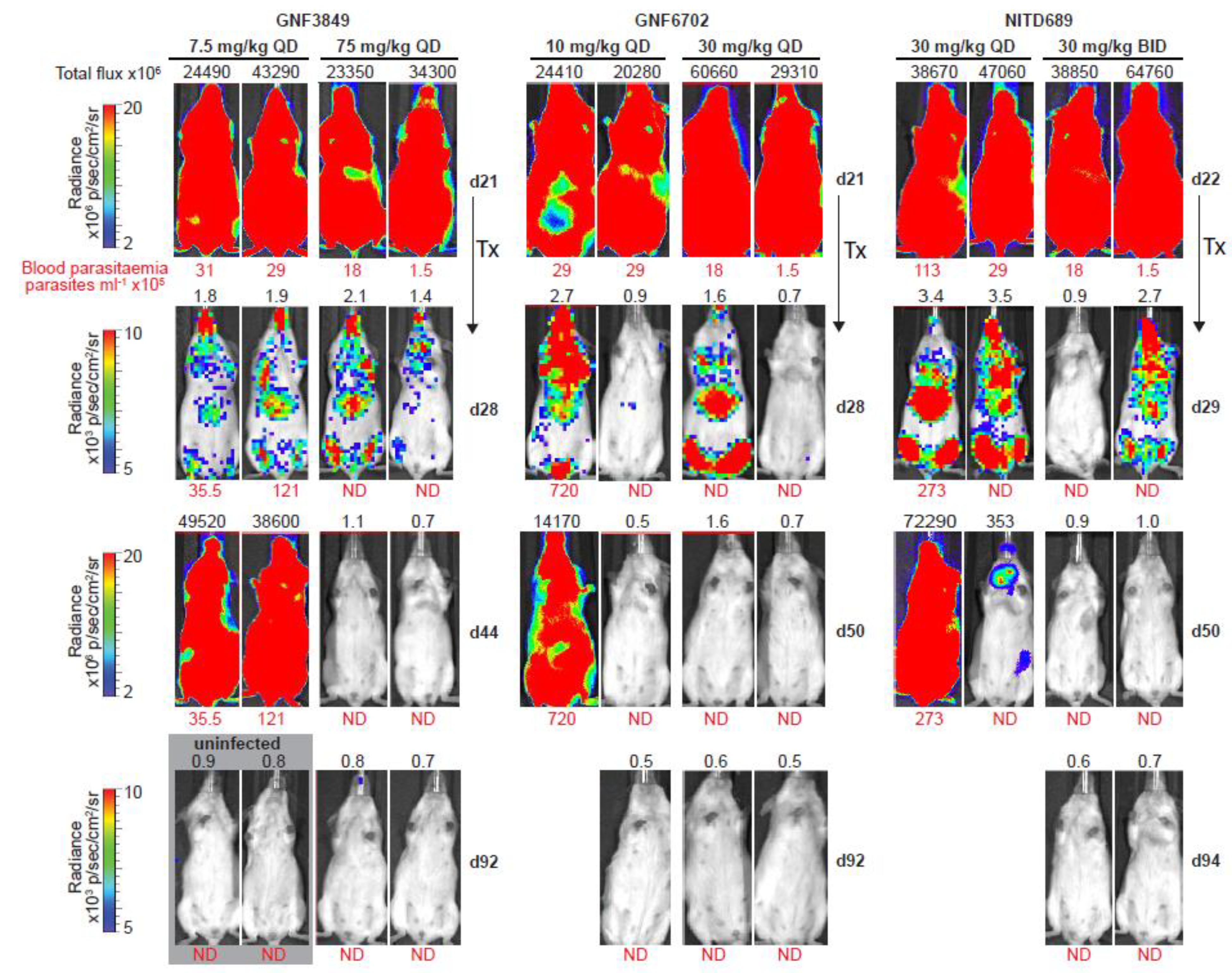
3.8. TP Class Compounds Are Efficacious against Meningoencephalic Infection in HAT Mouse Model
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. World Health Organization: Human African Trypanosomiasis; WHO: Rome, Italy, 2019. [Google Scholar]
- Yun, O.; Priotto, G.; Tong, J.; Flevaud, L.; Chappuis, F. Nect is next: Implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e720. [Google Scholar] [CrossRef] [PubMed]
- Drugs for Neglected Diseases initiative. Sleeping Sickness: Current Treatments; Drugs for Neglected Diseases initiative: Geneva, Switzerland, 2019. [Google Scholar]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [PubMed]
- Trindade, S.; Rijo-Ferreira, F.; Carvalho, T.; Pinto-Neves, D.; Guegan, F.; Aresta-Branco, F.; Bento, F.; Young, S.A.; Pinto, A.; Van Den Abbeele, J.; et al. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe 2016, 19, 837–848. [Google Scholar] [CrossRef]
- Capewell, P.; Cren-Travaille, C.; Marchesi, F.; Johnston, P.; Clucas, C.; Benson, R.A.; Gorman, T.A.; Calvo-Alvarez, E.; Crouzols, A.; Jouvion, G.; et al. The skin is a significant but overlooked anatomical reservoir for vector-borne african trypanosomes. Elife 2016, 5, e17716. [Google Scholar] [CrossRef] [PubMed]
- Buscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.; Myburgh, E.; Gao, M.Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, L.; Tilton, S.; Wang, J. Development of a high throughput equilibrium solubility assay using miniaturized shake-flask method in early drug discovery. J. Pharm. Sci. 2007, 96, 3052–3071. [Google Scholar] [CrossRef]
- Faller, B. Artificial membrane assays to assess permeability. Curr. Drug Metab. 2008, 9, 886–892. [Google Scholar] [CrossRef]
- Waters, N.J.; Jones, R.; Williams, G.; Sohal, B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J. Pharm. Sci. 2008, 97, 4586–4595. [Google Scholar] [CrossRef]
- Kalvass, J.C.; Tess, D.A.; Giragossian, C.; Linhares, M.C.; Maurer, T.S. Influence of microsomal concentration on apparent intrinsic clearance: Implications for scaling in vitro data. Drug Metab. Dispos. 2001, 29, 1332–1336. [Google Scholar] [PubMed]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. Scyx-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Myburgh, E.; Coles, J.A.; Ritchie, R.; Kennedy, P.G.; McLatchie, A.P.; Rodgers, J.; Taylor, M.C.; Barrett, M.P.; Brewer, J.M.; Mottram, J.C. In vivo imaging of trypanosome-brain interactions and development of a rapid screening test for drugs against cns stage trypanosomiasis. PLoS Negl. Trop. Dis. 2013, 7, e2384. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, S.C.; Nerima, B.; Maser, P.; Brun, R. Melarsoprol- and pentamidine-resistant Trypanosoma brucei rhodesiense populations and their cross-resistance. Int. J. Parasitol. 2007, 37, 1443–1448. [Google Scholar] [CrossRef]
- Graf, F.E.; Ludin, P.; Arquint, C.; Schmidt, R.S.; Schaub, N.; Kunz Renggli, C.; Munday, J.C.; Krezdorn, J.; Baker, N.; Horn, D.; et al. Comparative genomics of drug resistance in Trypanosoma brucei rhodesiense. Cell Mol. Life Sci. 2016, 73, 3387–3400. [Google Scholar] [CrossRef]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef]
- Diaz, R.; Luengo-Arratta, S.A.; Seixas, J.D.; Amata, E.; Devine, W.; Cordon-Obras, C.; Rojas-Barros, D.I.; Jimenez, E.; Ortega, F.; Crouch, S.; et al. Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Negl. Trop. Dis. 2014, 8, e3253. [Google Scholar] [CrossRef]
- Cleghorn, L.A.; Albrecht, S.; Stojanovski, L.; Simeons, F.R.; Norval, S.; Kime, R.; Collie, I.T.; De Rycker, M.; Campbell, L.; Hallyburton, I.; et al. Discovery of indoline-2-carboxamide derivatives as a new class of brain-penetrant inhibitors of Trypanosoma brucei. J. Med. Chem. 2015, 58, 7695–7706. [Google Scholar] [CrossRef]
- Wyllie, S.; Brand, S.; Thomas, M.; De Rycker, M.; Chung, C.W.; Pena, I.; Bingham, R.P.; Bueren-Calabuig, J.A.; Cantizani, J.; Cebrian, D.; et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 9318–9323. [Google Scholar] [CrossRef]
- De Rycker, M.; O’Neill, S.; Joshi, D.; Campbell, L.; Gray, D.W.; Fairlamb, A.H. A static-cidal assay for Trypanosoma brucei to aid hit prioritisation for progression into drug discovery programmes. PLoS Negl. Trop. Dis. 2012, 6, e1932. [Google Scholar] [CrossRef][Green Version]
- Nagendar, P.; Gillespie, J.R.; Herbst, Z.M.; Ranade, R.M.; Molasky, N.M.R.; Faghih, O.; Turner, R.M.; Gelb, M.H.; Buckner, F.S. Triazolopyrimidines and imidazopyridines: Structure-activity relationships and in vivo efficacy for trypanosomiasis. ACS Med. Chem. Lett. 2019, 10, 105–110. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assays/Properties * | GNF3849 # | GNF6702 # | NITD689 $ |
|---|---|---|---|
| T. b. brucei EC50 (nM) | 22 ± 4 | 70 ± 3 | 30 ± 4 |
| HepG2 CC50 (µM) | 1.1 ± 0.2 | >20 | >20 |
| Solubility pH 6.8 (g/L) | <0.002 | 0.009 | 0.071 |
| LogD/Mol Wt/PSA | 3.31/428/98 | 2.32/429/103 | 3.05/408/98 |
| MPO | 3.4 | 3.7 | 3.6 |
| PAMPA Permeability (% FA) | 96 | 92 | 99.1 |
| Mouse liver microsomal clearance (µL/min/mg) | 15.2 | 34.1 | 7.41 |
| Mouse plasma protein binding (%) | >99 | 95.1 | 90.5 |
| Parasite | Strain | GNF3849 | GNF6702 | Melarsoprol | Pentamidine |
|---|---|---|---|---|---|
| T. b. gambiense (EC50 in nM) | STIB930 | 4.5 ± 1 | 3.7 ± 2 | 7.1 ± 2.7 | 1.4 ± 0.9 |
| K048 | 4.8 ± 1.1 | 3.5 ± 1.4 | 10.2 ± 1.9 | 33.9 ± 16 | |
| R130 | 1.9 ± 1 | 2.3 ± 1.1 | 7.7 ± 3.1 | 13.5 ± 4 | |
| T. b. rhodesiense (EC50 in nM) | STIB900 | 1.4 ± 1 | 1.6 ± 0.9 | 3.9 ± 2 | 2.1 ± 0.7 |
| STIB900 PentR | 1.7 ± 1 | 1.2 ± 0.3 | 46.8 ± 19 | 203 ± 54 | |
| STIB900 MelR | 0.6 ± 3 | 0.7 ± 0.2 | 95.5 ± 24 | 217 ± 86 | |
| T. b. brucei (EC50 in nM) | BS221 | 1.9 ± 0.8 | 0.9 ± 0.3 | 4.7 ± 1.8 | 0.3 ± 0.1 |
| BS221 (P2 KO) | 4.0 ± 2.5 | 3.5 ± 1.2 | 37.7 ± 14.8 | 3.1 ± 0.4 | |
| STIB950 | 0.9 ± 0.2 | 1.2 ± 0.5 | 16.9 ± 8.8 | 0.5 ± 0.2 |
| Strain | T. brucei PSMB4WT (EC50 = nM) | T. brucei PSMB4F24L (EC50 = nM) |
|---|---|---|
| GNF3849 | 10 ± 3 | 2200 ± 20 |
| GNF6702 | 18 ± 1.8 | 1200 ± 13 |
| NITD689 | 20 ± 4 | 1900 ± 20 |
| Bortezomib | 0.94 ± 0.05 | 1.1 ± 0.26 |
| Parameters | Units | GNF3849 | GNF6702 | NITD689 |
|---|---|---|---|---|
| I.V. PK | ||||
| Dose | mg/kg | 5 | 5 | 5 |
| Vss | L/kg | 1.2 | 1.2 | 1.5 |
| CL | mL/min/kg | 2.46 | 2.26 | 13.48 |
| T1/2 | H | 6.5 | 7.0 | 1.6 |
| P.O. PK | ||||
| Dose | mg/kg | 20 | 20 | 20 |
| Cmax | nM | 7160 (<71.6) | 13,668 (669.7) | 12,170 (1156.2) |
| Tmax | H | 3 | 7.67 | 0.5 |
| AUC | nM*h | 107,510 (<1075.1) | 271,489 (13,303) | 89,692 (8235.7) |
| F | % | 34 | 79 | 100 |
| Compound ID | Dose (mg/kg) | Dose Frequency | Mice Cured/Total | Mean day of Relapse | % Cured |
|---|---|---|---|---|---|
| GNF3849 | 7.5 | QD | 6/6 | >31 | 100 |
| 25 | QD | 6/6 | >31 | 100 | |
| 75 | QD | 6/6 | >31 | 100 | |
| GNF6702 | 0.1 | QD | 0/6 | 6.83 | 0 |
| 0.3 | QD | 2/6 | 19 | 33.3 | |
| 1 | QD | 6/6 | >31 | 100 | |
| 3 | QD | 6/6 | >31 | 100 | |
| 10 | QD | 6/6 | >31 | 100 | |
| NITD689 | 1 | QD | 0/6 | 19.5 | 0 |
| 1.5 | BID | 1/6 | 21 | 16.7 | |
| 3 | QD | 3/6 | 21 | 50 | |
| 10 | QD | 6/6 | >31 | 100 |
| Compound ID | BTB (%) | MDR1-MDCK | Efflux Ratio | B/P Ratio | ||
|---|---|---|---|---|---|---|
| A–B | B–A | B–A/A–B | 5 min | 60 min | ||
| GNF3849 | >99 | 4.5 | 14.1 | 3.2 | 0.22 | 0.28 |
| GNF6702 | 94.7 | 8.3 | 22.1 | 2.7 | 0.13 | 0.17 |
| NITD689 | 96.5 | 32.6 | 29.2 | 0.9 | 0.12 | 0.12 |
| Compound ID | Dose (mg/kg) | Dose Frequency | Mice Cured/Total | Mean Day of Relapse | % Cured |
|---|---|---|---|---|---|
| GNF3849 | 7.5 | QD | 0/6 | 37 | 0 |
| 75 | QD | 3/6 | 60 (>92 *) | 50 | |
| GNF6702 | 3 | QD | 0/6 | 47 | 0 |
| 10 | QD | 2/6 | 50 (>92 *) | 66 | |
| 30 | QD | 6/6 | >92 | 100 | |
| 100 | QD | 6/6 | >92 | 100 | |
| NITD689 | 15 | BID | 0/6 | 42 | 0 |
| 30 | QD | 0/6 | 50 | 0 | |
| 30 | BID | 6/6 | >94 | 100 | |
| 60 | QD | 6/6 | >94 | 100 | |
| Diminazene | 40 | QD | 0/3 | 42 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, S.P.S.; Lakshminarayana, S.B.; Jiricek, J.; Kaiser, M.; Ritchie, R.; Myburgh, E.; Supek, F.; Tuntland, T.; Nagle, A.; Molteni, V.; et al. Anti-Trypanosomal Proteasome Inhibitors Cure Hemolymphatic and Meningoencephalic Murine Infection Models of African Trypanosomiasis. Trop. Med. Infect. Dis. 2020, 5, 28. https://doi.org/10.3390/tropicalmed5010028
Rao SPS, Lakshminarayana SB, Jiricek J, Kaiser M, Ritchie R, Myburgh E, Supek F, Tuntland T, Nagle A, Molteni V, et al. Anti-Trypanosomal Proteasome Inhibitors Cure Hemolymphatic and Meningoencephalic Murine Infection Models of African Trypanosomiasis. Tropical Medicine and Infectious Disease. 2020; 5(1):28. https://doi.org/10.3390/tropicalmed5010028
Chicago/Turabian StyleRao, Srinivasa P S, Suresh B Lakshminarayana, Jan Jiricek, Marcel Kaiser, Ryan Ritchie, Elmarie Myburgh, Frantisek Supek, Tove Tuntland, Advait Nagle, Valentina Molteni, and et al. 2020. "Anti-Trypanosomal Proteasome Inhibitors Cure Hemolymphatic and Meningoencephalic Murine Infection Models of African Trypanosomiasis" Tropical Medicine and Infectious Disease 5, no. 1: 28. https://doi.org/10.3390/tropicalmed5010028
APA StyleRao, S. P. S., Lakshminarayana, S. B., Jiricek, J., Kaiser, M., Ritchie, R., Myburgh, E., Supek, F., Tuntland, T., Nagle, A., Molteni, V., Mäser, P., Mottram, J. C., Barrett, M. P., & Diagana, T. T. (2020). Anti-Trypanosomal Proteasome Inhibitors Cure Hemolymphatic and Meningoencephalic Murine Infection Models of African Trypanosomiasis. Tropical Medicine and Infectious Disease, 5(1), 28. https://doi.org/10.3390/tropicalmed5010028