First Draft Genome of the Trypanosomatid Herpetomonas muscarum ingenoplastis through MinION Oxford Nanopore Technology and Illumina Sequencing

 , , ,
, , ,  , , , ,
, , , ,  and
and 
{kind=link}
Abstract
1. Introduction
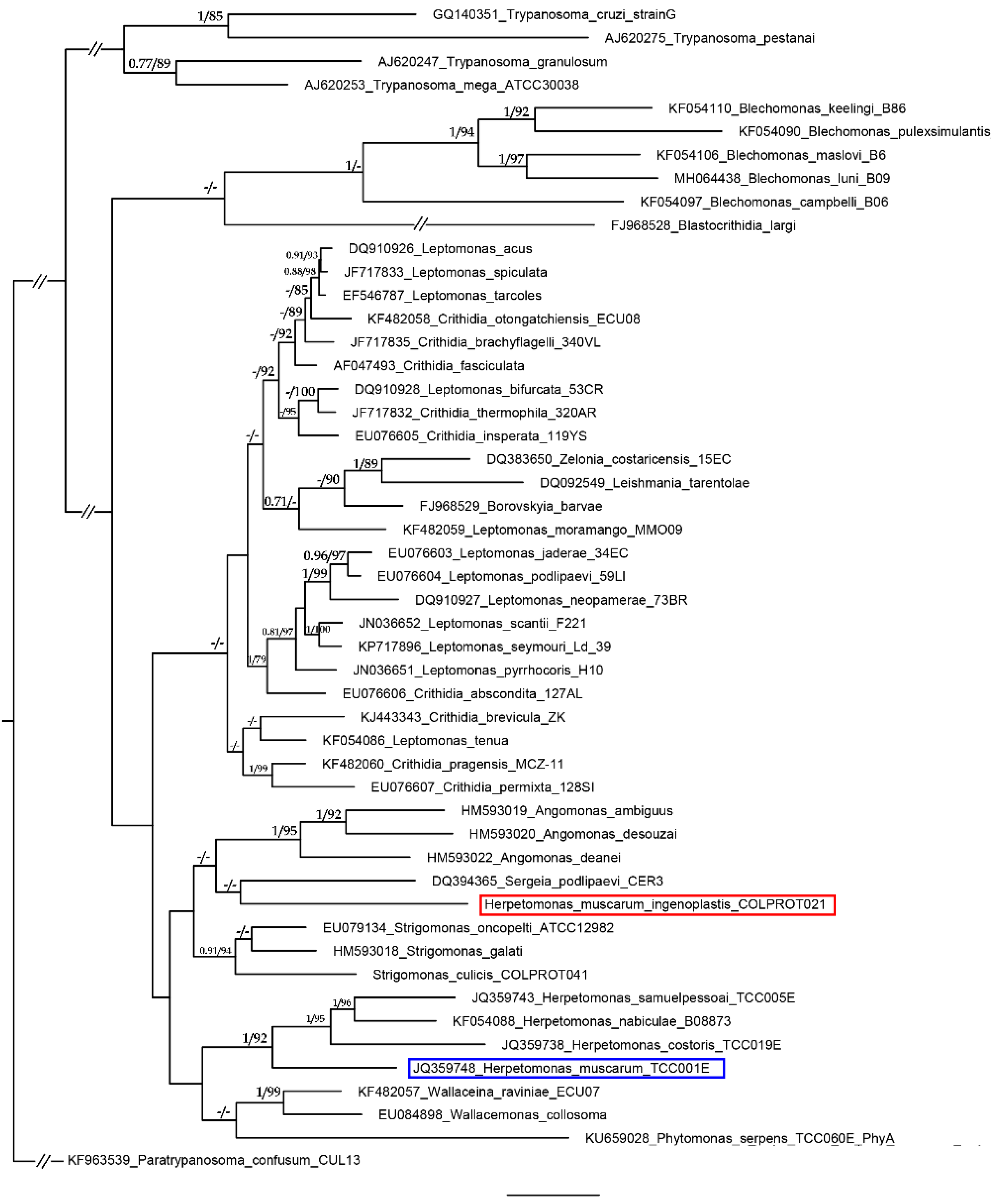
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Lukeš, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votýpka, J.; Yurchenko, V. Trypanosomatids are much more than just trypanosomes: Clues from the expanded family tree. Trends Parasitol. 2019, 34, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, K.; Honek, J.; Cadmus, C.M.; Efferth, T. Trypanosomatid parasites causing neglected diseases. Curr. Med. Chem. 2010, 17, 1594–1617. [Google Scholar] [CrossRef] [PubMed]
- Sangenito, L.S.; da Silva Santos, V.; d’Avila-Levy, C.M.; Branquinha, M.H.; Santos, A.L.S.; Oliveira, S.S.C. Leishmaniasis and Chagas Disease—Neglected Tropical Diseases: Treatment Updates. Curr. Top. Med. Chem. 2019, 19, 174–177. [Google Scholar] [CrossRef]
- Podlipaev, S.A. Insect trypanosomatids: The need to know more. Mem. Inst. Oswaldo Cruz 2000, 95, 517–522. [Google Scholar] [CrossRef]
- Maslov, D.A.; Votýpka, J.; Yurchenko, V.; Lukeš, J. Diversity and phylogeny of insect trypanosomatids: All that is hidden shall be revealed. Trends Parasitol. 2013, 29, 43–52. [Google Scholar] [CrossRef]
- d’Avila-Levy, C.M.; Boucinha, C.; Kostygov, A.; Santos, H.L.; Morelli, K.A.; Grybchuk-Ieremenko, A.; Duval, L.; Votýpka, J.; Yurchenko, V.; Grellier, P.; et al. Exploring the environmental diversity of kinetoplastid flagellates in the high-throughput DNA sequencing era. Mem. Inst. Oswaldo Cruz 2015, 110, 956–965. [Google Scholar] [CrossRef]
- Rogers, W.E.; Wallace, F.G. Two new subspecies of Herpetomonus muscurum (Leidy, 1856) Kent, 1880. J. Protozool. 1971, 18, 645–649. [Google Scholar] [CrossRef]
- Putnam, R.J. The role of carrion-frequenting arthropods in the decay process. Ecol. Entomol. 1978, 3, 133–139. [Google Scholar] [CrossRef]
- Votýpka, J.; Klepetková, H.; Jirků, M.; Kment, P.; Lukeš, J. Phylogenetic relationships of trypanosomatids parasitising true bugs (Insecta: Heteroptera) in sub-Saharan Africa. Int. J. Parasitol. 2012, 42, 489–500. [Google Scholar] [CrossRef]
- Týč, J.; Votýpka, J.; Klepetková, H.; Suláková, H.; Jirků, M.; Lukeš, J. Growing diversity of trypanosomatid parasites of flies (Diptera: Brachycera): Frequent cosmopolitism and moderate host specificity. Mol. Phylogenet. Evol. 2013, 69, 255–264. [Google Scholar] [CrossRef]
- Ishemgulova, A.; Butenko, A.; Kortišová, L.; Boucinha, C.; Grybchuk-Ieremenko, A.; Morelli, K.A.; Tesařová, M.; Kraeva, N.; Grybchuk, D.; Pánek, T.; et al. Molecular mechanisms of thermal resistance of the insect trypanosomatid Crithidia thermophila. PLoS ONE 2017, 12, e0174165. [Google Scholar] [CrossRef] [PubMed]
- Brunoro, G.V.F.; Menna-Barreto, R.F.S.; Garcia-Gomes, A.S.; Boucinha, C.; Lima, D.B.; Carvalho, P.C.; Teixeira-Ferreira, A.; Trugilho, M.R.O.; Perales, J.; Schwämmle, V.; et al. Quantitative Proteomic Map of the Trypanosomatid Strigomonas culicis: The Biological Contribution of its Endosymbiotic Bacterium. Protist 2019, 170, 125698. [Google Scholar] [CrossRef] [PubMed]
- Camargo, E.P. Phytomonas and other trypanosomatid parasites of plants and fruit. Adv. Parasitol. 1999, 42, 29–112. [Google Scholar] [PubMed]
- d’Avila-Levy, C.M.; Yurchenko, V.; Votýpka, J.; Grellier, P. Protist Collections: Essential for Future Research. Trends Parasitol. 2016, 32, 840–842. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Mikheenko, A.; Valin, G.; Prjibelski, A.; Saveliev, V.; Gurevich, A. Icarus: Visualizer for de novo assembly evaluation. Bioinformatics 2016, 32, 3321–3323. [Google Scholar] [CrossRef]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Helaers, R.; Milinkovitch, M.C. MetaPIGA v2.0: Maximum likelihood large phylogeny estimation using the metapopulation genetic algorithm and other stochastic heuristics. BMC Bioinform. 2010, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
d’Avila-Levy, C.M.; Bearzatto, B.; Ambroise, J.; Helaers, R.; Butenko, A.; Yurchenko, V.; A. Morelli, K.; L. C. Santos, H.; Brouillard, P.; Grellier, P.; et al. First Draft Genome of the Trypanosomatid Herpetomonas muscarum ingenoplastis through MinION Oxford Nanopore Technology and Illumina Sequencing. Trop. Med. Infect. Dis. 2020, 5, 25. https://doi.org/10.3390/tropicalmed5010025
d’Avila-Levy CM, Bearzatto B, Ambroise J, Helaers R, Butenko A, Yurchenko V, A. Morelli K, L. C. Santos H, Brouillard P, Grellier P, et al. First Draft Genome of the Trypanosomatid Herpetomonas muscarum ingenoplastis through MinION Oxford Nanopore Technology and Illumina Sequencing. Tropical Medicine and Infectious Disease. 2020; 5(1):25. https://doi.org/10.3390/tropicalmed5010025
Chicago/Turabian Styled’Avila-Levy, Claudia M., Bertrand Bearzatto, Jérôme Ambroise, Raphaël Helaers, Anzhelika Butenko, Vyacheslav Yurchenko, Karina A. Morelli, Helena L. C. Santos, Pascal Brouillard, Philippe Grellier, and et al. 2020. "First Draft Genome of the Trypanosomatid Herpetomonas muscarum ingenoplastis through MinION Oxford Nanopore Technology and Illumina Sequencing" Tropical Medicine and Infectious Disease 5, no. 1: 25. https://doi.org/10.3390/tropicalmed5010025
APA Styled’Avila-Levy, C. M., Bearzatto, B., Ambroise, J., Helaers, R., Butenko, A., Yurchenko, V., A. Morelli, K., L. C. Santos, H., Brouillard, P., Grellier, P., Gala, J.-L., & Vikkula, M. (2020). First Draft Genome of the Trypanosomatid Herpetomonas muscarum ingenoplastis through MinION Oxford Nanopore Technology and Illumina Sequencing. Tropical Medicine and Infectious Disease, 5(1), 25. https://doi.org/10.3390/tropicalmed5010025