Phenotypic Drug Discovery for Human African Trypanosomiasis: A Powerful Approach

 ,
,
Abstract
1. Introduction
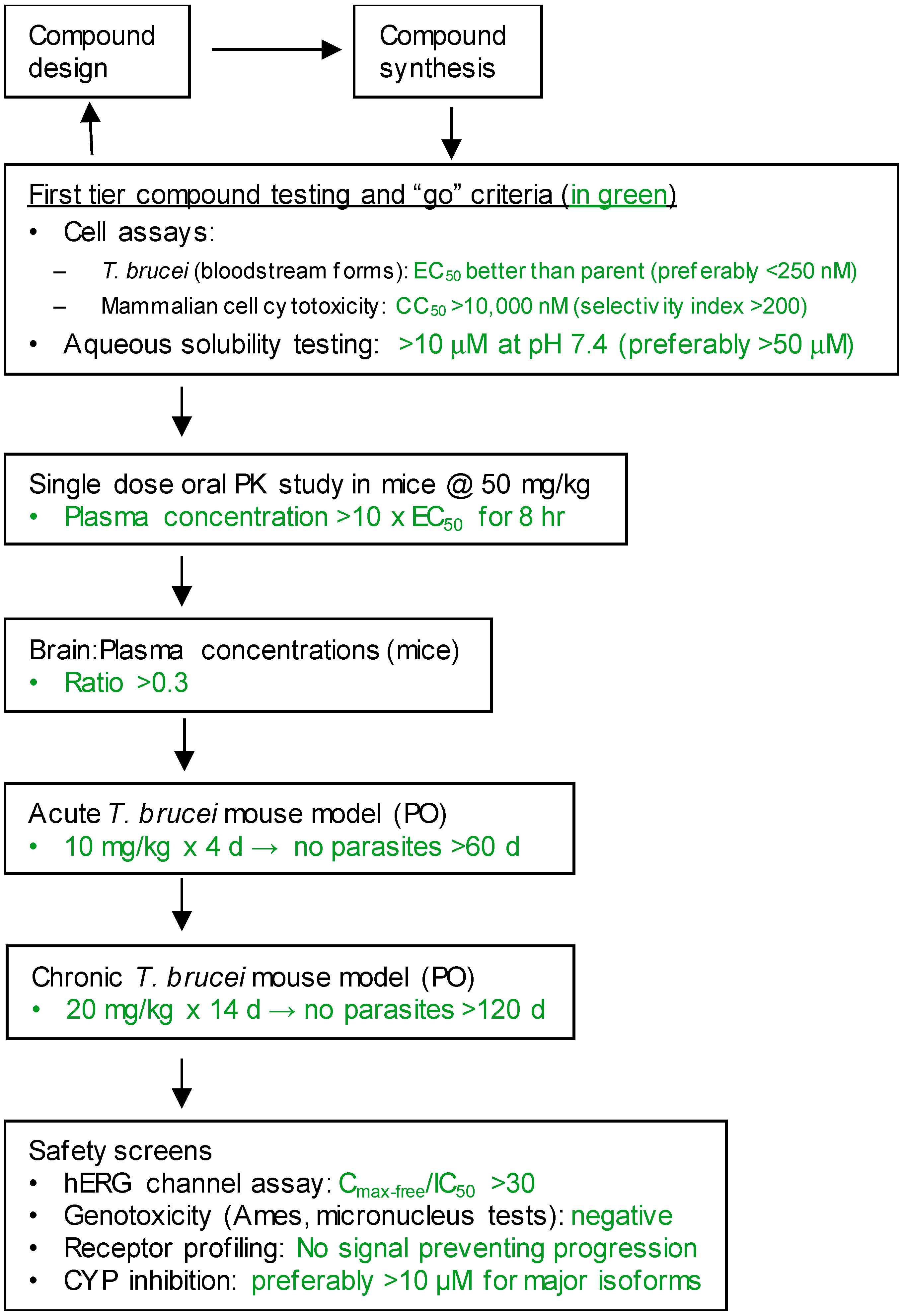
2. Materials and Methods
2.1. Phenotypic Screen for T. brucei Growth Arrest
2.2. In Vitro Parasite Growth Arrest Assay
2.3. Mammalian Cell Cytotoxicity Assay
2.4. Solubility Measurement
2.5. Permeability Across Monolayers of MDCKII-MDR1 Cells
2.6. Pharmacokinetic Studies in Mice
2.7. Brain Permeability Studies
2.8. Anti-Parasite Efficacy Studies in Mice (Acute Model)
2.9. Anti-Parasite Efficacy Studies in Mice (Chronic Model)
2.10. Chemical Synthesis Procedures
2.11. Metabolite Identification
3. Results
3.1. Selection of Hit Compounds
3.2. Screening for Brain Permeability
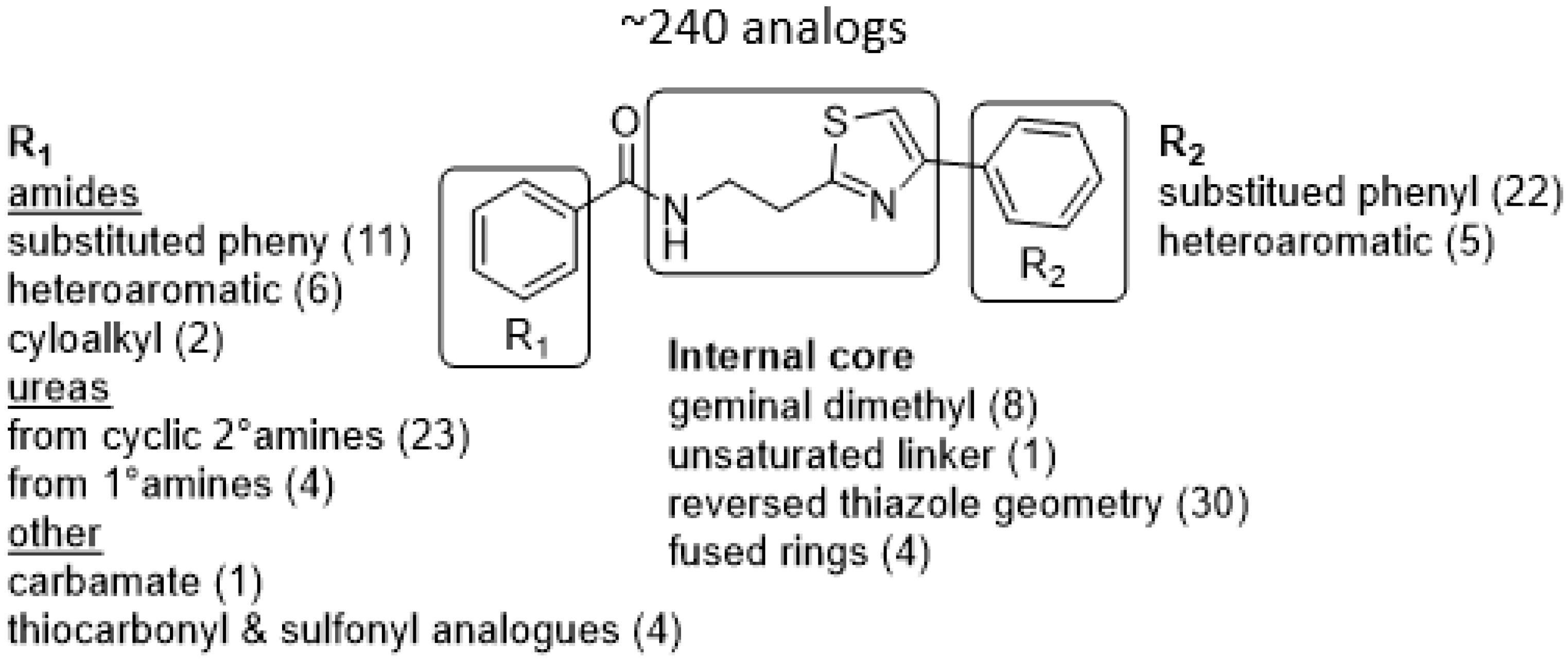
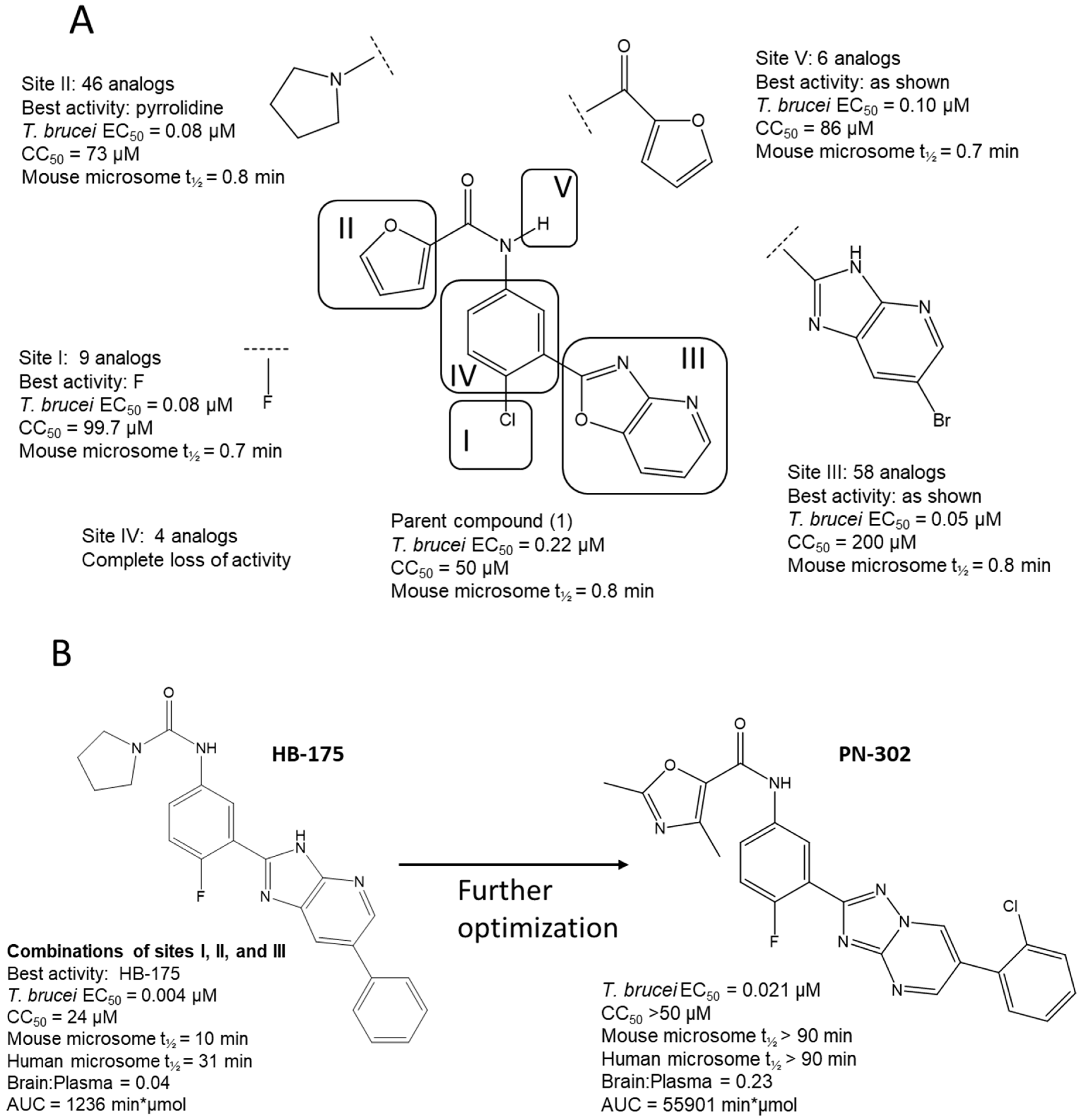
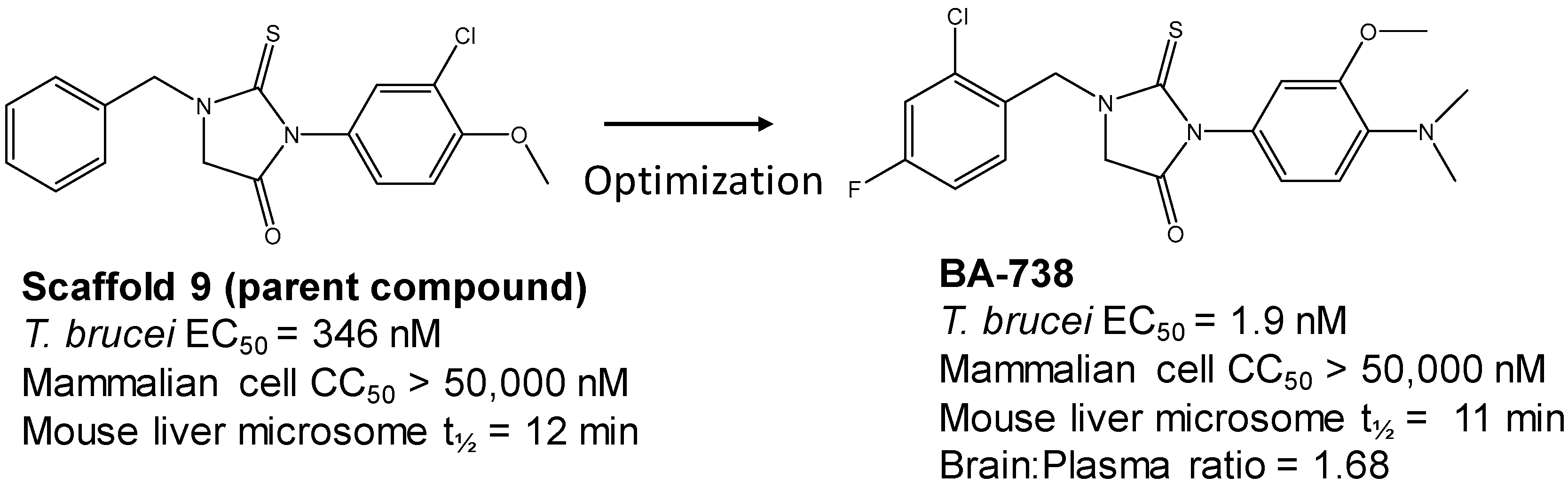
3.3. Hit-to-Lead Optimization
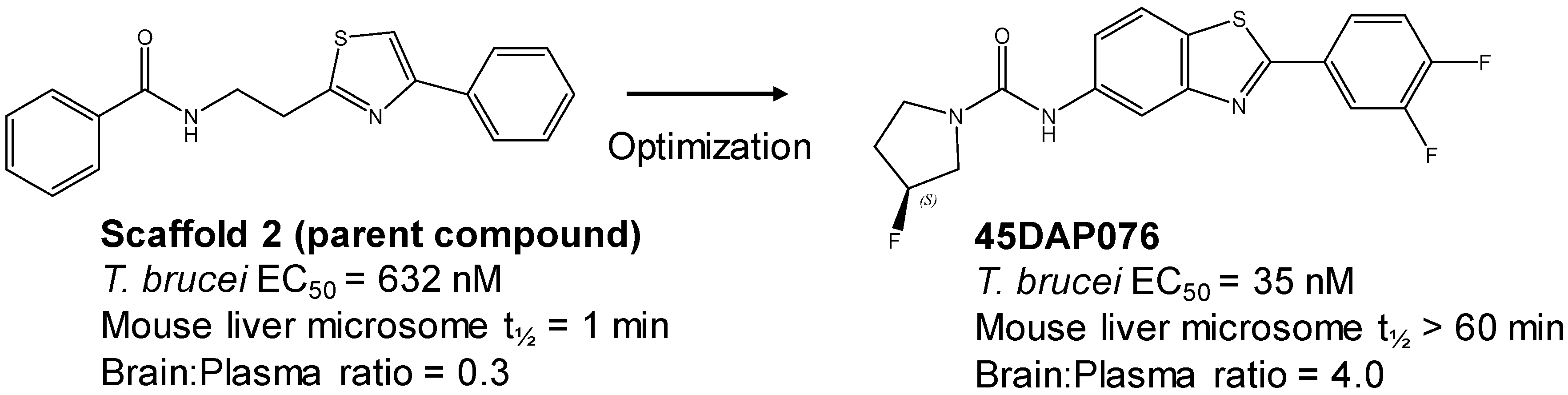
3.4. Active Compound Series (Highlights)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Lutje, V.; Seixas, J.; Kennedy, A. Chemotherapy for second-stage Human African trypanosomiasis. Cochrane Database Syst. Rev. 2013, 6, CD006201. [Google Scholar] [CrossRef]
- WHO Interim Guidelines for the Treatment of Gambiense Human African Trypanosomiasis. Available online: https://apps.who.int/iris/handle/10665/326178 (accessed on 4 February 2020).
- DNDi. HAT Target Product Profile. Available online: https://www.dndi.org/diseases-projects/hat/hat-target-product-profile/ (accessed on 4 February 2020).
- Tatipaka, H.B.; Gillespie, J.R.; Chatterjee, A.K.; Norcross, N.R.; Hulverson, M.A.; Ranade, R.M.; Nagendar, P.; Creason, S.A.; McQueen, J.; Duster, N.A.; et al. Substituted 2-phenylimidazopyridines: A new class of drug leads for human African trypanosomiasis. J. Med. Chem. 2014, 57, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Cnubben, N.H.; Wijnholds, J.; Van Deemter, L.; Van Bladeren, P.J.; Borst, P. Transport of glutathione prostaglandin A conjugates by the multidrug resistance protein 1. FEBS Lett. 1997, 419, 112–116. [Google Scholar] [CrossRef]
- Suryadevara, P.K.; Olepu, S.; Lockman, J.W.; Ohkanda, J.; Karimi, M.; Verlinde, C.L.; Kraus, J.M.; Schoepe, J.; Van Voorhis, W.C.; Hamilton, A.D.; et al. Structurally simple inhibitors of lanosterol 14alpha-demethylase are efficacious in a rodent model of acute Chagas disease. J. Med. Chem. 2009, 52, 3703–3715. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.A.; Gillespie, J.R.; McQueen, J.; Hulverson, M.A.; Ranade, R.M.; Creason, S.A.; Herbst, Z.M.; Gelb, M.H.; Buckner, F.S.; Tidwell, R.R. Urea derivatives of 2-aryl-benzothiazol-5-amines: A new class of potential drugs for human African trypanosomiasis. J. Med. Chem. 2017, 60, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Macherey, A.C.; Dansette, P. Chemical mechanisms of toxicity: Basic knowledge for designing safer drugs. In The Practice of Medicinal Chemistry, 2nd ed.; Wermuth, C.G., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2003; pp. 545–560. [Google Scholar]
- Buchynskyy, A.G.; Gillespie, J.R.; Herbst, Z.; Ranade, R.; Buckner, F.S.; Gelb, M.H. 1-Benzyl-3-aryl-2-thiohydantoin derivatives as anti-Trypanosoma brucei agents: SAR and in-vivo efficacy. ACS Med. Chem. Lett. 2017, 8, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. The Practice of Medicinal Chemistry, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Nagendar, P.; Gillespie, J.R.; Herbst, Z.M.; Ranade, R.M.; Molasky, N.M.R.; Faghih, O.; Turner, R.M.; Gelb, M.H.; Buckner, F.S. Triazolopyrimidines and imidazopyridines as antitrypanosomal agents: Structure-activity relationships and in vivo efficacy. ACS Med. Chem. Lett. 2018, 10, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.A.; Wenzler, T.; Yang, S.; Weiser, P.T.; Wang, M.Z.; Brun, R.; Tidwell, R.R. Synthesis of novel amide and urea derivatives of thiazol-2-ethylamines and their activity against Trypanosoma brucei rhodesiense. Bioorg. Med. Chem. 2016, 24, 2451–2465. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.G.; Gillespie, J.R.; Ranade, R.M.; Herbst, Z.M.; Nguyen, U.T.T.; Buckner, F.S.; Montanari, C.A.; Gelb, M.H. New class of antitrypanosomal agents based on imidazopyridines. ACS Med. Chem. Lett. 2017, 8, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Buchynskyy, A.; Gillespie, J.R.; Hulverson, M.A.; McQueen, J.; Creason, S.A.; Ranade, R.M.; Duster, N.A.; Gelb, M.H.; Buckner, F.S. Discovery of N-(2-aminoethyl)-N-benzyloxyphenyl benzamides: New potent Trypanosoma brucei inhibitors. Bioorg. Med. Chem. 2017, 25, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.; Myburgh, E.; Gao, M.Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Brand, S.; Thomas, M.; De Rycker, M.; Chung, C.W.; Pena, I.; Bingham, R.P.; Bueren-Calabuig, J.A.; Cantizani, J.; Cebrian, D.; et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 9318–9323. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold | Structure | MW (g/mol) | Clog P | T. brucei EC50 (nM) | Mammalian CC50 (nM) * | B/P Ratio (Mouse) ** |
|---|---|---|---|---|---|---|
| a. | ||||||
| 1 |  | 339.7 | 3.21 | 217.6 | >50,000 | 0.547 |
| 2 |  | 308.4 | 3.77 | 480 | >50,000 | 0.3 |
| 3 |  | 349.4 | 3.56 | 873.7 | 37,968 | 2.86 |
| 4 |  | 363.6 | 3.48 | 790 | 15,860 | 2.00 |
| 5 |  | 443.4 | 3.69 | 657.5 | >100,000 | 0.845 |
| 6 |  | 328.4 | 2.02 | 306.9 | >100,000 | 0.727 |
| 7 |  | 468.9 | 2.87 | 656.6 | >50,000 | 0.587 |
| 8 |  | 294.3 | 2.4 | 214.4 | 14,300 | 7.11 |
| 9 |  | 346.8 | 3.72 | 346 | >50,000 | 0.743 *** |
| b. | ||||||
| 10 |  | 428.5 | 3.67 | 2438 | 0.224 | |
| 11 |  | 413.376 | 5.11 | 3089 | 0.09 | |
| 12 |  | 438.54 | 4.17 | 1700 | >100,000 | 0.267 |
| 13 |  | 391.49 | 2.66 | 1000 | >100,000 | 0.02 |
| 14 |  | 510.565 | 5.5 | 707.5 | >100,000 | 0.01 |
| 15 |  | 505.618 | 8.15 | 1500 | 33,000 | 0 |
| 16 |  | 487.57 | 6.37 | 650 | >100,000 | No plasma exposure |
| 17 |  | 465.57 | 6.17 | 3500 | >100,000 | 0 |
| Compound Series | # Analogs Made | Status | Reason for Discontinuation | Reference |
|---|---|---|---|---|
| 1 | 253 | Active | [5,14] | |
| 2 | 249 | Active | [8,15,16] | |
| 3 | 131 | Stopped | Insufficient improvement in EC50 | |
| 4 | 141 | Stopped | Slow killing activity (“static”) | [17] |
| 5 | 102 | Stopped | Poor solubility. Poor PK. | |
| 6 | 41 | Stopped | Insufficient improvement in EC50 | |
| 7 | 138 | Stopped | Poor metabolic stability; Poor PK | |
| 8 | 91 | Stopped | Insufficient improvement in EC50 | |
| 9 | 280 | Active | [11] | |
| 10 | 66 | Stopped | Insufficient improvement in EC50 | |
| 11 | 47 | Stopped | Insufficient improvement in EC50 | |
| 12 | 0 | Not started | ||
| Total | 1539 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckner, F.S.; Buchynskyy, A.; Nagendar, P.; Patrick, D.A.; Gillespie, J.R.; Herbst, Z.; Tidwell, R.R.; Gelb, M.H. Phenotypic Drug Discovery for Human African Trypanosomiasis: A Powerful Approach. Trop. Med. Infect. Dis. 2020, 5, 23. https://doi.org/10.3390/tropicalmed5010023
Buckner FS, Buchynskyy A, Nagendar P, Patrick DA, Gillespie JR, Herbst Z, Tidwell RR, Gelb MH. Phenotypic Drug Discovery for Human African Trypanosomiasis: A Powerful Approach. Tropical Medicine and Infectious Disease. 2020; 5(1):23. https://doi.org/10.3390/tropicalmed5010023
Chicago/Turabian StyleBuckner, Frederick S., Andriy Buchynskyy, Pendem Nagendar, Donald A. Patrick, J. Robert Gillespie, Zackary Herbst, Richard R. Tidwell, and Michael H. Gelb. 2020. "Phenotypic Drug Discovery for Human African Trypanosomiasis: A Powerful Approach" Tropical Medicine and Infectious Disease 5, no. 1: 23. https://doi.org/10.3390/tropicalmed5010023
APA StyleBuckner, F. S., Buchynskyy, A., Nagendar, P., Patrick, D. A., Gillespie, J. R., Herbst, Z., Tidwell, R. R., & Gelb, M. H. (2020). Phenotypic Drug Discovery for Human African Trypanosomiasis: A Powerful Approach. Tropical Medicine and Infectious Disease, 5(1), 23. https://doi.org/10.3390/tropicalmed5010023