Paramyxo- and Coronaviruses in Rwandan Bats

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods:
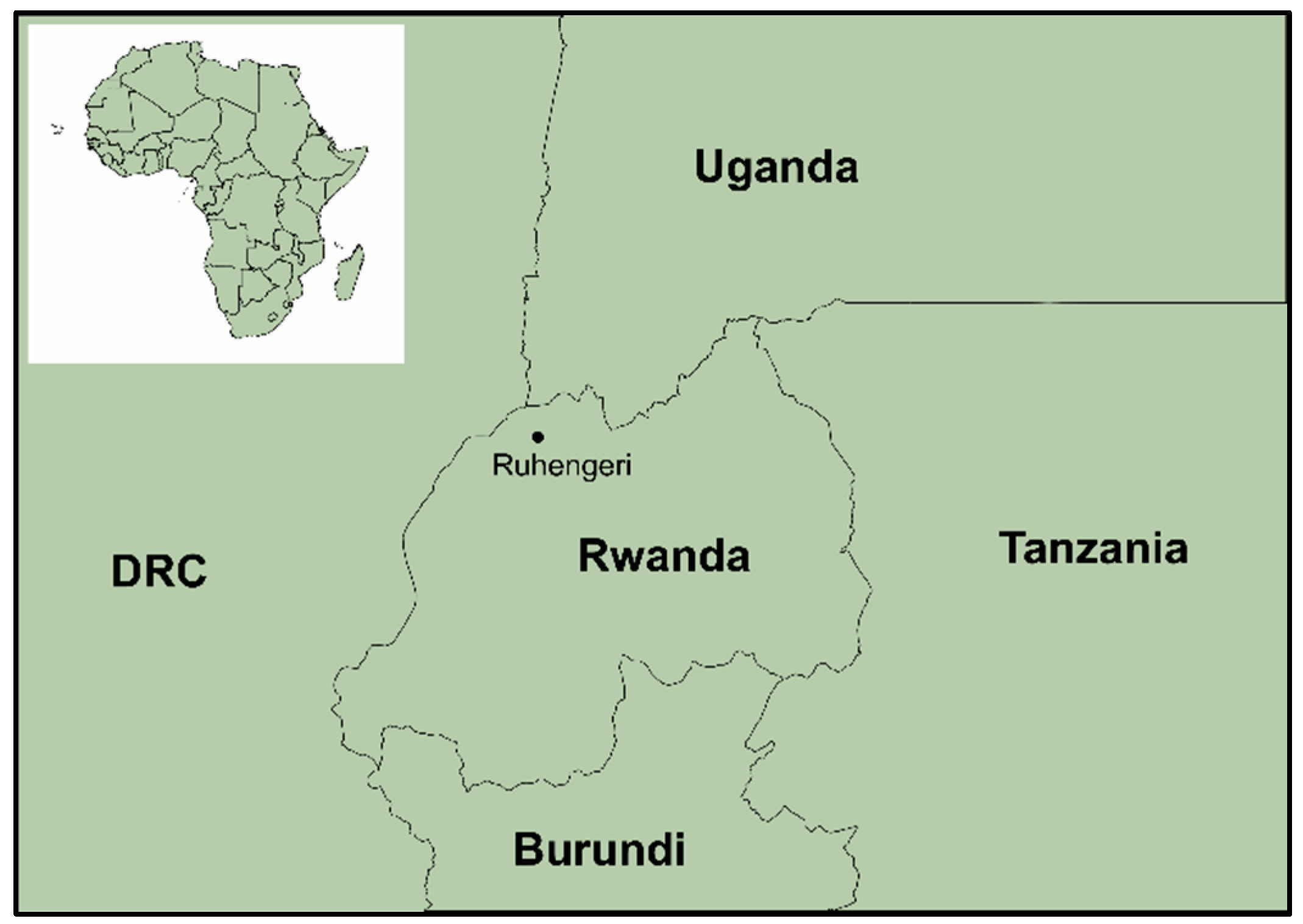
2.1. Study Area and Sample Collection
2.2. RNA and DNA Extraction
2.3. Morphological and Molecular Host Species Identification
2.4. PCR Amplification of Viral Targets
2.4.1. Coronaviruses
2.4.2. Paramyxoviruses
2.5. Sequencing and Phylogenetic Analysis
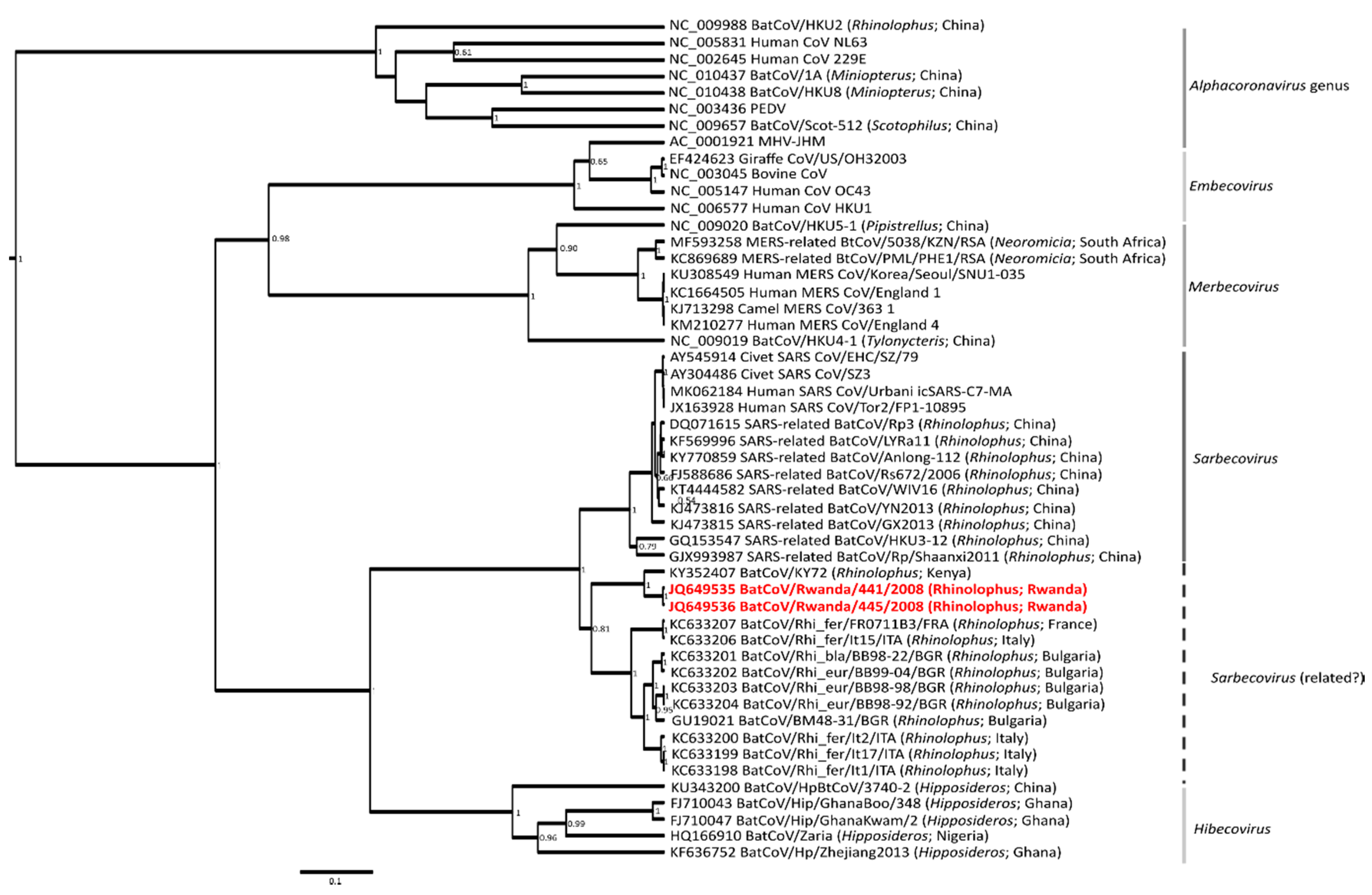
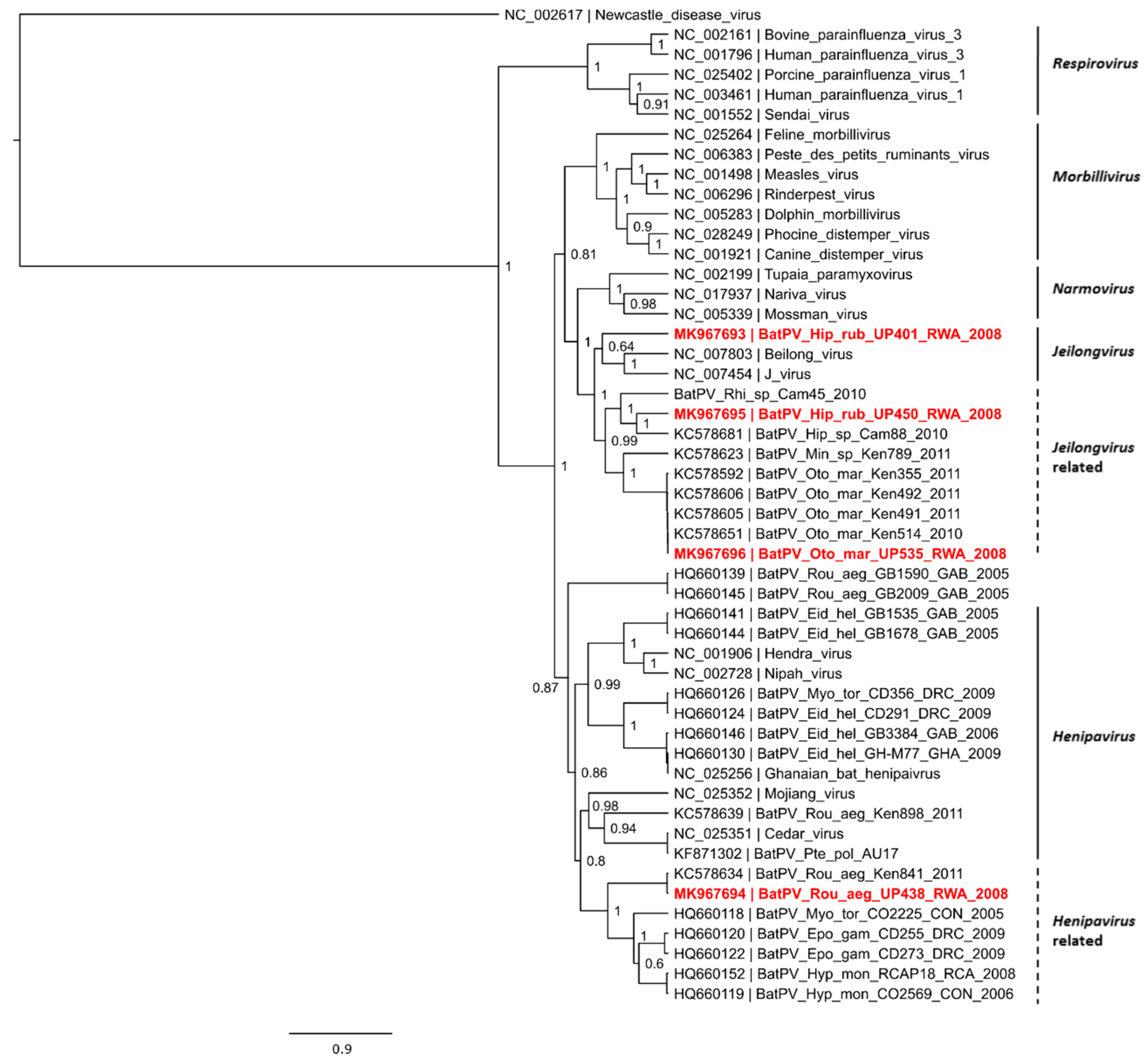
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hayman, D.T.S. Filoviruses in bats: current knowledge and future directions. Viruses 2014, 6, 1759–1788. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Markotter, W.; Coertse, J. Bat lyssaviruses. Rev. Sci. Tech. OIE 2018, 37, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.H.; Brewer, T.F.; Madoff, L.C.; Pollack, M.P.; Sonricker, A.L.; Keller, M.; Freifeld, C.C.; Blench, M.; Mawudeku, A.; Brownstein, J.S. Global capacity for emerging infectious disease detection. Proc. Natl. Acad. Sci. USA 2010, 107, 21701–21706. [Google Scholar] [CrossRef] [PubMed]
- Daszak, P.; Cunningham, A.A.; Hyatt, A.D. Emerging Infectious Diseases of Wildlife-- Threats to Biodiversity and Human Health. Science 2000, 287, 443–449. [Google Scholar] [CrossRef]
- Emperador, D.M.; Mazzola, L.T.; Wonderly Trainor, B.; Chua, A.; Kelly-Cirino, C. Diagnostics for filovirus detection: impact of recent outbreaks on the diagnostic landscape. BMJ Glob. Heal. 2019, 4, e001112. [Google Scholar] [CrossRef]
- Amman, B.R.; Albarino, C.G.; Bird, B.H.; Nyakarahuka, L.; Sealy, T.K.; Balinandi, S.; Schuh, A.J.; Campbell, S.M.; Stroher, U.; Jones, M.E.B.; et al. A recently discovered pathogenic paramyxovirus, Sosuga virus, is present in Rousettus aegyptiacus fruit bats at multiple locations in Uganda. J. Wildl. Dis. 2015, 51, 774–779. [Google Scholar] [CrossRef]
- Peel, A.J.; Sargan, D.R.; Baker, K.S.; Hayman, D.T.S.; Barr, J.A.; Crameri, G.; Suu-Ire, R.; Broder, C.C.; Lembo, T.; Wang, L.F.; et al. Continent-wide panmixia of an African fruit bat facilitates transmission of potentially zoonotic viruses. Nat. Commun. 2013, 4, 1–14. [Google Scholar] [CrossRef]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 2018, 58, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, 1–28. [Google Scholar] [CrossRef]
- Ge, X.; Li, J.; Yang, X.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and Characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Yang, X.-L.; Hu, B.; Wang, B.; Wang, M.-N.; Zhang, Q.; Zhang, W.; Wu, L.-J.; Ge, X.-Y.; Zhang, Y.-Z.; Daszak, P.; et al. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of Severe Acute Respiratory Syndrome coronavirus. J. Virol. 2016, 90, 3253–3256. [Google Scholar] [CrossRef]
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482–485. [Google Scholar] [CrossRef]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio 2010, 1, e00208-10–e00208-18. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, M.; Weyer, J.; Nel, L.H.; Markotter, W. Coronaviruses in South African bats. Vector Borne Zoonotic Dis. 2013, 13, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History. J. Virol. 2017, 91, e01953–e01956. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further evidence for bats as the evolutionary source of Middle East respiratory syndrome coronavirus. MBio 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, M.; Mortlock, M.; Weyer, J.; Bezuidt, O.; Seamark, E.; Kearney, T.; Gleasner, C.; Erkkila, T.; Cui, H.; Markotter, W. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE 2018, 13, e0194527. [Google Scholar] [CrossRef]
- Virtue, E.R.; Marsh, G.A.; Wang, L.-F. Paramyxoviruses infecting humans: the old, the new and the unknown. Future Microbiol. 2009, 4, 537–554. [Google Scholar] [CrossRef]
- ICTV Virus Taxonomy: 2018b Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 1 May 2019).
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef]
- Chua, K.B.; Goh, K.J.; Wong, K.T.; Kamarulzaman, A.; Seow, P.; Tan, K.; Ksiazek, T.G.; Zaki, S.R.; Paul, G.; Lam, S.K.; et al. Fatal encephalitis due to Nipah virus among pig farmers in Malaysia. Lancet 1999, 354, 1257–1259. [Google Scholar] [CrossRef]
- Albariño, C.G.; Foltzer, M.; Towner, J.S.; Rowe, L.A.; Campbell, S.; Jaramillo, C.M.; Bird, B.H.; Reeder, D.M.; Vodzak, M.E.; Rota, P.; et al. Novel paramyxovirus associated with severe acute febrile disease, South Sudan and Uganda, 2012. Emerg. Infect. Dis. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Young, P.L.; Field, H.E.; Mackenzie, J.S. Isolation of Hendra virus from pteropid bats: A natural reservoir of Hendra virus. J. Gen. Virol. 2000, 81, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B.; Koh, C.L.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Baker, K.S.; Todd, S.; Marsh, G.; Wood, J.L.N.; Wang, L.F.; Murcia, P.R.; Cunningham, A.A. Co-circulation of diverse paramyxoviruses in an urban African fruit bat population. J. Gen. Virol. 2012, 93, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, M.; Kuzmin, I.V.; Weyer, J.; Gilbert, A.T.; Agwanda, B.; Rupprecht, C.E.; Nel, L.H.; Kearney, T.; Malekani, J.M.; Markotter, W. Novel paramyxoviruses in bats from sub-Saharan Africa, 2007–2012. Emerg. Infect. Dis. 2015, 21, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, M.; Dietrich, M.; Weyer, J.; Paweska, J.T.; Markotter, W. Co-circulation and excretion dynamics of diverse rubula-and related viruses in Egyptian rousette bats from South Africa. Viruses 2019, 11, 37. [Google Scholar] [CrossRef]
- Van Cakenberghe, V.; Gembu Tungaluna, G.-C.; Musaba Akawa, P.; Seamark, E.; Verheyen, E. The bats of the Congo and of Rwanda and Burundi revisited (Mammalia: Chiroptera). Eur. J. Taxon. 2017, 1–327. [Google Scholar] [CrossRef]
- Greenberg, J.A.; Dimenna, M.A.; Hanelt, B.; Hofkin, B.V. Analysis of post-blood meal flight distances in mosquitoes utilizing zoo animal blood meals. J. Vector Ecol. 2012, 37, 83–89. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Lassen, S.B.; Nielsen, S.A.; Skovgård, H.; Kristensen, M. Molecular identification of bloodmeals from biting midges (Diptera: Ceratopogonidae: Culicoides Latreille) in Denmark. Parasitol. Res. 2011, 108, 823–829. [Google Scholar] [CrossRef]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and Broadly Reactive Reverse Transcription-PCR Assays to Detect Novel Paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Hall, T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: more models, new heuristics and high- performance computing. Nat. Methods 2015, 9, 6–9. [Google Scholar]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Hu, Z.; Zhang, H.; Zhang, J.; Eaton, B.T.; Zhang, S.; et al. Bats are natural reservoirs of SARS-Like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Mélade, J.; Wieseke, N.; Ramasindrazana, B.; Flores, O.; Lagadec, E.; Gomard, Y.; Goodman, S.M.; Dellagi, K.; Pascalis, H. An eco-epidemiological study of Morbilli-related paramyxovirus infection in Madagascar bats reveals host-switching as the dominant macro-evolutionary mechanism. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Willoughby, A.R.; Phelps, K.L.; Phelps, K.L.; Predict Consortium; Olival, K.J. A comparative analysis of viral richness and viral sharing in cave-roosting bats. Diversity 2017, 9, 35. [Google Scholar] [CrossRef]
- Kurth, A.; Kohl, C.; Brinkmann, A.; Ebinger, A.; Harper, J.A.; Wang, L.-F.; Mühldorfer, K.; Wibbelt, G. Novel paramyxoviruses in free-ranging European bats. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Vidgen, M.E.; de Jong, C.; Rose, K.; Hall, J.; Field, H.E.; Smith, C.S. Novel paramyxoviruses in Australian flying-fox populations support host-virus coevolution. J. Gen. Virol. 2015, 96, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.A.; Temmam, S.; Lebarbenchon, C.; Lagadec, E.; Chotte, J.; Guillebaud, J.; Ramasindrazana, B.; Héraud, J.-M.; de Lamballerie, X.; Goodman, S.M.; et al. Identification of novel paramyxoviruses in insectivorous bats of the Southwest Indian Ocean. Virus Res. 2012, 170, 159–163. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Bat Species | Paramyxovirus Positive/Total | Coronavirus Positive/Total | Ruhengeri Site |
|---|---|---|---|
| Hipposideros ruber (Noack’s roundleaf bat) | 2/3 | 0/2 | Cave 1 |
| Otomops martiensseni (Large eared free-tailed bat) | 1/4 | 0/15 | Cave 1 |
| Rousettus aegyptiacus (Egyptian Rousette fruit bat) | 1/14 | 0/72 | Cave 1 |
| Rhinolophus spp. (Horseshoe bat) | 0/2 | 2/7 | Cave 2 |
| Epomophorus spp. (Epauletted fruit bat) | - | 0/5 | Cave 1 |
| Total | 4/23 | 2/101 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markotter, W.; Geldenhuys, M.; Jansen van Vuren, P.; Kemp, A.; Mortlock, M.; Mudakikwa, A.; Nel, L.; Nziza, J.; Paweska, J.; Weyer, J. Paramyxo- and Coronaviruses in Rwandan Bats. Trop. Med. Infect. Dis. 2019, 4, 99. https://doi.org/10.3390/tropicalmed4030099
Markotter W, Geldenhuys M, Jansen van Vuren P, Kemp A, Mortlock M, Mudakikwa A, Nel L, Nziza J, Paweska J, Weyer J. Paramyxo- and Coronaviruses in Rwandan Bats. Tropical Medicine and Infectious Disease. 2019; 4(3):99. https://doi.org/10.3390/tropicalmed4030099
Chicago/Turabian StyleMarkotter, Wanda, Marike Geldenhuys, Petrus Jansen van Vuren, Alan Kemp, Marinda Mortlock, Antoine Mudakikwa, Louis Nel, Julius Nziza, Janusz Paweska, and Jacqueline Weyer. 2019. "Paramyxo- and Coronaviruses in Rwandan Bats" Tropical Medicine and Infectious Disease 4, no. 3: 99. https://doi.org/10.3390/tropicalmed4030099
APA StyleMarkotter, W., Geldenhuys, M., Jansen van Vuren, P., Kemp, A., Mortlock, M., Mudakikwa, A., Nel, L., Nziza, J., Paweska, J., & Weyer, J. (2019). Paramyxo- and Coronaviruses in Rwandan Bats. Tropical Medicine and Infectious Disease, 4(3), 99. https://doi.org/10.3390/tropicalmed4030099