
Comprehensive Pharmacological Management of Wilson’s Disease: Mechanisms, Clinical Strategies, and Emerging Therapeutic Innovations


Abstract

1. Introduction
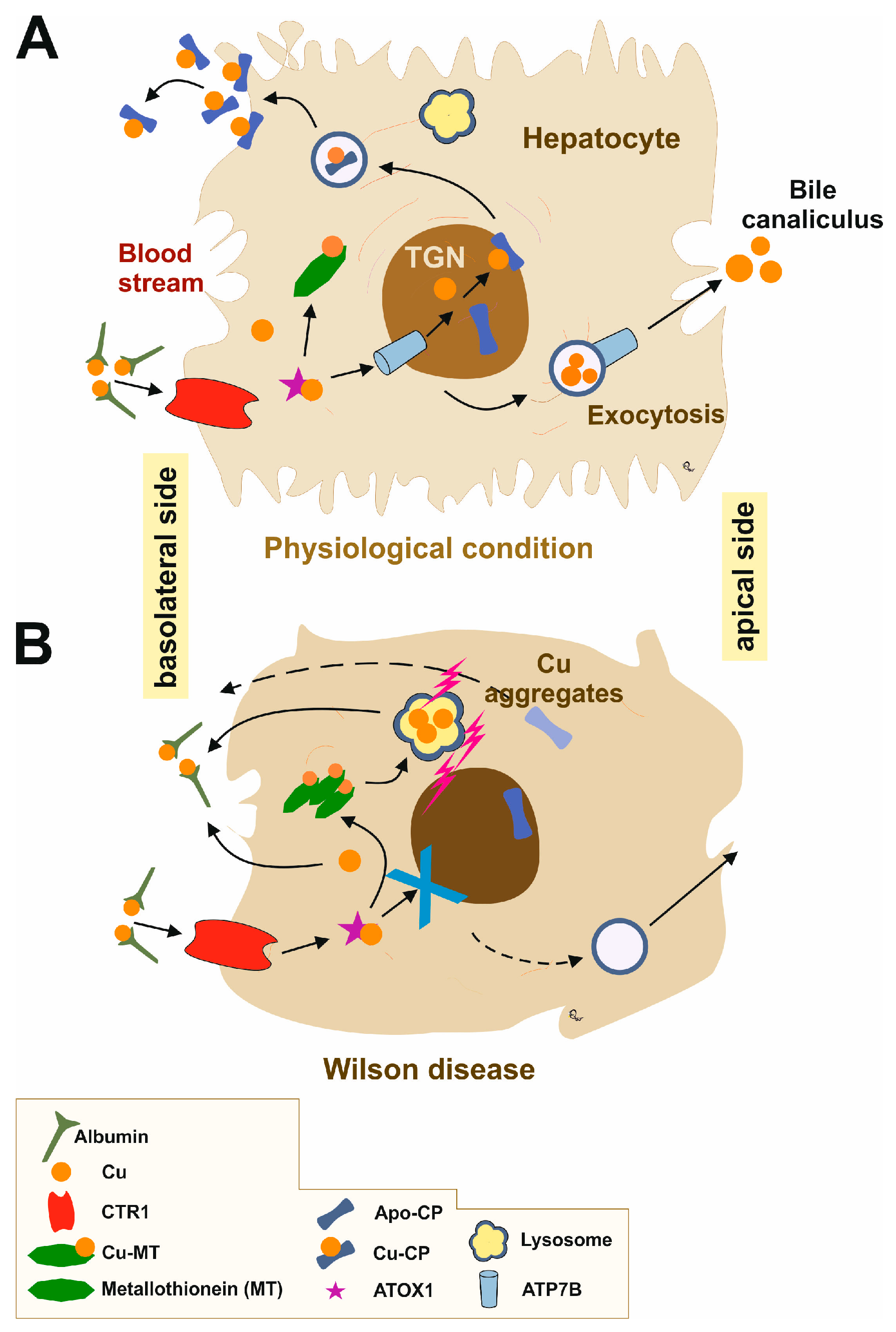
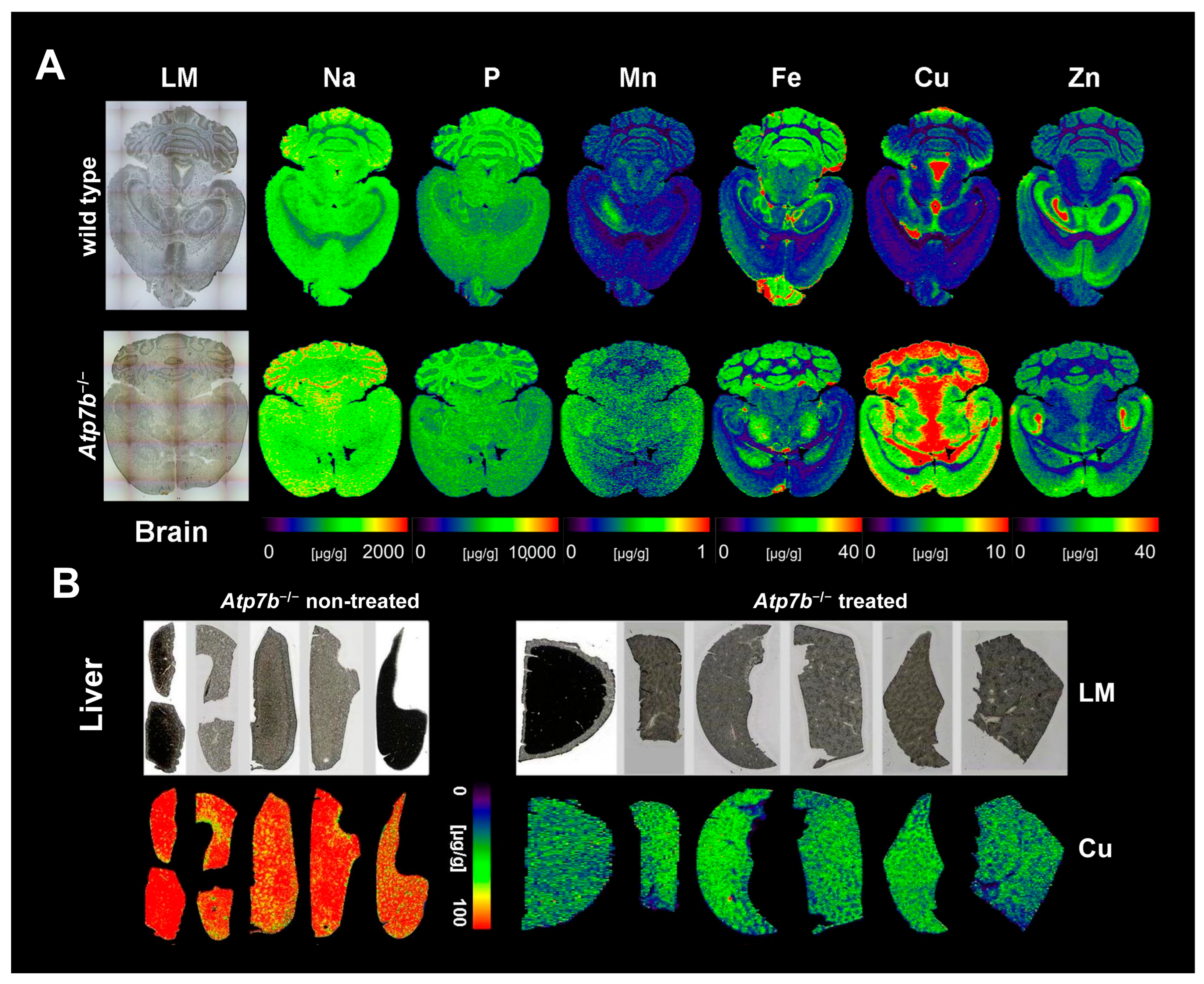
2. Overview of Wilson’s Disease Pathophysiology
3. Pharmacological Management
3.1. British Anti-Lewisite
3.2. D-Penicillamine
3.3. Trientine
3.4. Tetrathiomolybdate
3.5. Dimcercaptosuccinic Acid
3.6. Ethylenediaminetetraacetic Acid
3.7. Bis-Choline Tetrathiomolybdate
3.8. Zinc
3.9. Summary of Pharmacological Agents
3.10. Combination Therapy with Antioxidants and Anti-Inflammatory
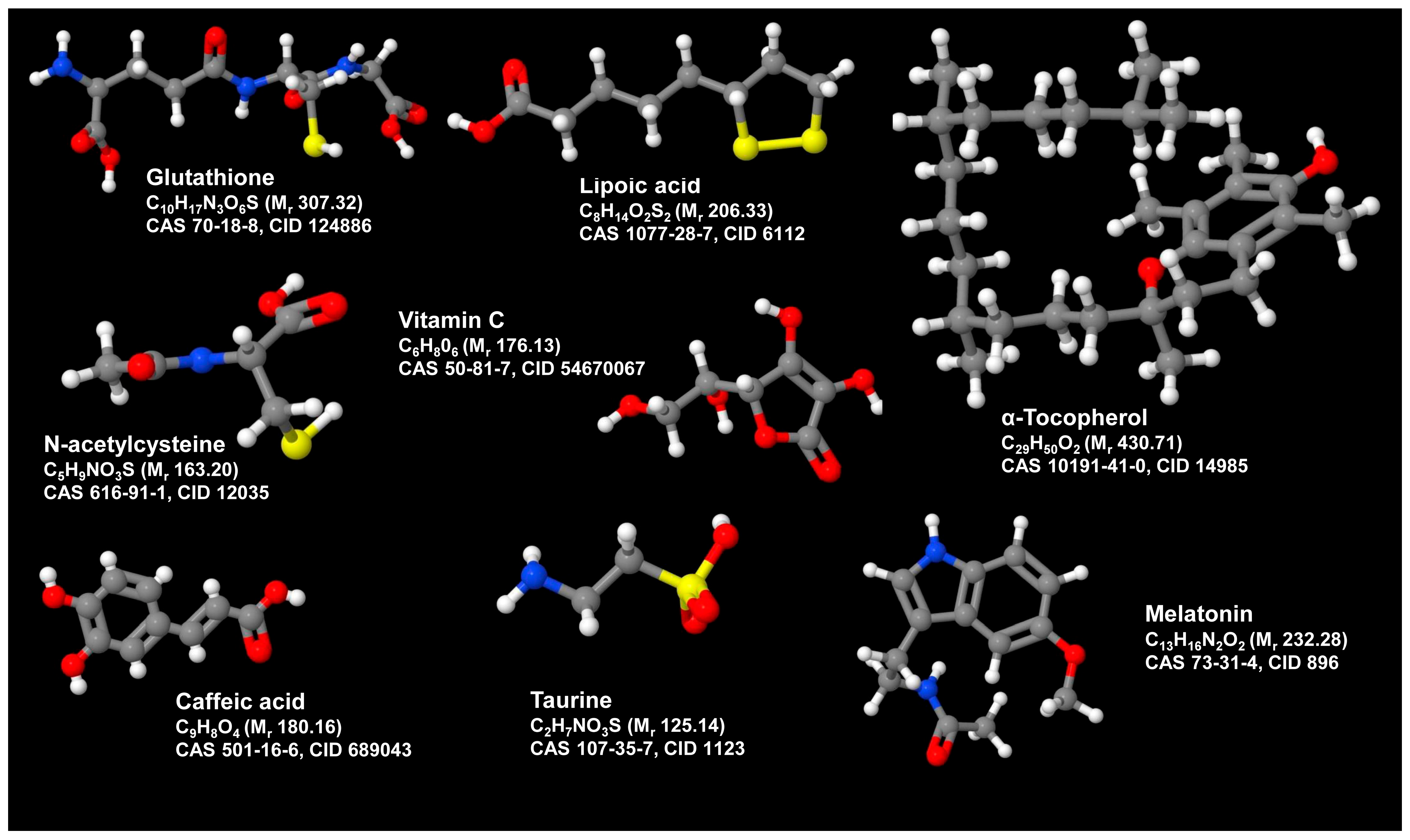
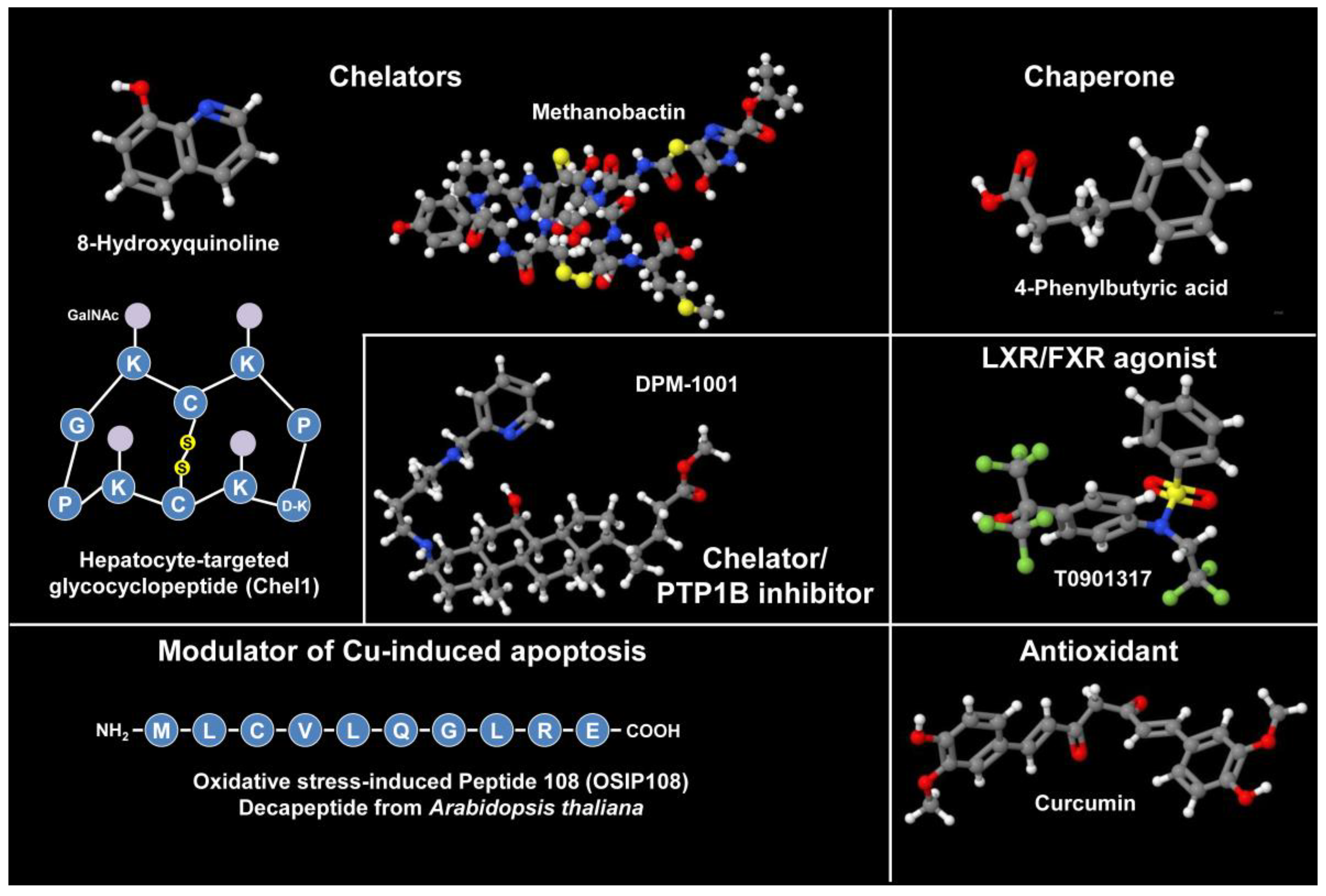
3.11. Emerging/Adjunct Therapies
4. Monitoring and Follow-Up
5. Guidelines and Recommendations
5.1. General Guidelines
5.2. Management Considerations for Pediatric Patients
6. Future Directions
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AASLD | American Association for the Study of Liver Diseases |
| ATP7B | ATPase, Cu2+-transporting, beta polypeptide |
| BAL | British anti-lewisite |
| DMSA | Dimercaptosuccinic acid |
| EASL | European Association for the Study of the Liver |
| EDTA | Ethylenediaminetetraacetic acid |
| ROS | Reactive oxygen species |
| TETA | Triethylenetetramine |
| TTM | Tetrathiomolybdate |
| WDA | Wilson Disease Association |
| WDSG-UK | Wilson’s Disease Support Group UK |
References
- Penning, L.C.; Berenguer, M.; Czlonkowska, A.; Double, K.L.; Dusek, P.; Espinós, C.; Lutsenko, S.; Medici, V.; Papenthin, W.; Stremmel, W.; et al. A Century of progress on Wilson disease and the enduring challenges of genetics, diagnosis, and treatment. Biomedicines 2023, 11, 420. [Google Scholar] [CrossRef]
- Dooley, J.S. The history of Wilson disease. Clin. Liver Dis. 2024, 23, e0238. [Google Scholar] [CrossRef]
- Stremmel, W.; Weiskirchen, R. An initiative to promote the integration of artificial intelligence in transforming the diagnosis and management of Wilson disease in the 21st century. Metab. Target. Organ. Damage 2025, 5, 9. [Google Scholar] [CrossRef]
- Schilsky, M.L.; Roberts, E.A.; Bronstein, J.M.; Dhawan, A.; Hamilton, J.P.; Rivard, A.M.; Washington, M.K.; Weiss, K.H.; Zimbrean, P.C. A multidisciplinary approach to the diagnosis and management of Wilson disease: Executive summary of the 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology 2023, 77, 1428–1455. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL-ERN Clinical Practice Guidelines on Wilson’s disease. J. Hepatol. 2025, 82, 690–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wu, L.; Jiang, P.; Ji, Z.; Yin, M.; Zhou, H.; He, L.; Xia, Z.; Wang, F.; Xiao, X.; et al. Evaluation of salivary ceruloplasmin for the diagnosis of Wilson’s disease. Sci. Rep. 2025, 15, 8197. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health Office of Dietary Supplements. Available online: https://ods.od.nih.gov/factsheets/Copper-HealthProfessional/ (accessed on 4 June 2025).
- Avan, A.; Członkowska, A.; Gaskin, S.; Granzotto, A.; Sensi, S.L.; Hoogenraad, T.U. The role of zinc in the treatment of Wilson’s disease. Int. J. Mol. Sci. 2022, 23, 9316. [Google Scholar] [CrossRef]
- Kalita, J.; Kumar, V.; Misra, U.K.; Parashar, V.; Ranjan, A. Adjunctive antioxidant therapy in neurologic Wilson’s disease improves the outcomes. J. Mol. Neurosci. 2020, 70, 378–385. [Google Scholar] [CrossRef]
- Zheng, Z.W.; Dong, Y.; Wu, Z.Y. Latest innovations in the treatment of Wilson’s disease. iLiver 2022, 1, 181–186. [Google Scholar] [CrossRef]
- Tang, S.; Bai, L.; Duan, Z.; Zheng, S. Stem cells treatment for Wilson disease. Curr. Stem Cell Res. Ther. 2022, 17, 712–719. [Google Scholar] [CrossRef]
- Wei, R.; Yang, J.; Cheng, C.W.; Ho, W.I.; Li, N.; Hu, Y.; Hong, X.; Fu, J.; Yang, B.; Liu, Y.; et al. CRISPR-targeted genome editing of human induced pluripotent stem cell-derived hepatocytes for the treatment of Wilson’s disease. JHEP Rep. 2021, 4, 100389. [Google Scholar] [CrossRef] [PubMed]
- Murillo, O.; Luqui, D.M.; Gazquez, C.; Martinez-Espartosa, D.; Navarro-Blasco, I.; Monreal, J.I.; Guembe, L.; Moreno-Cermeño, A.; Corrales, F.J.; Prieto, J.; et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J. Hepatol. 2016, 64, 419–426. [Google Scholar] [CrossRef]
- Uerlings, R.; Moreno, D.; Murillo, O.; Gazquez, C.; Hernández-Alcoceba, R.; González-Aseguinolaza, G.; Weiskirchen, R. Brain copper storage after genetic long-term correction in a mouse model of Wilson disease. Neurol. Genet. 2018, 4, e243. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.; Murillo, O.; Gazquez, C.; Hernandez-Alcoceba, R.; Uerlings, R.; Gonzalez-Aseguinolaza, G.; Weiskirchen, R. Visualization of the therapeutic efficacy of a gene correction approach in Wilson’s disease by laser-ablation inductively coupled mass spectrometry. J. Hepatol. 2018, 68, 1088–1090. [Google Scholar] [CrossRef]
- Choi, W.; Cha, S.; Kim, K. Navigating the CRISPR/Cas landscape for enhanced diagnosis and treatment of Wilson’s disease. Cells 2024, 13, 1214. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Monteagudo, A.; Ripollés, E.; Berenguer, M.; Espinós, C. Wilson’s disease: Facing the challenge of diagnosing a rare disease. Biomedicines 2021, 9, 1100. [Google Scholar] [CrossRef]
- Kumar, M.; Gaharwar, U.; Paul, S.; Poojary, M.; Pandhare, K.; Scaria, V.; Bk, B. WilsonGen a comprehensive clinically annotated genomic variant resource for Wilson’s Disease. Sci. Rep. 2020, 10, 9037. [Google Scholar] [CrossRef]
- Ovchinnikova, E.V.; Garbuz, M.M.; Ovchinnikova, A.A.; Kumeiko, V.V. Epidemiology of Wilson’s disease and pathogenic variants of the ATP7B gene leading to diversified protein disfunctions. Int. J. Mol. Sci. 2024, 25, 2402. [Google Scholar] [CrossRef]
- Sandahl, T.D.; Laursen, T.L.; Munk, D.E.; Vilstrup, H.; Weiss, K.H.; Ott, P. The prevalence of Wilson’s disease: An update. Hepatology 2020, 71, 722–732. [Google Scholar] [CrossRef]
- Chang, I.J.; Hahn, S.H. The genetics of Wilson disease. Handb. Clin. Neurol. 2017, 142, 19–34. [Google Scholar] [CrossRef]
- Chanpong, A.; Dhawan, A. Wilson disease in children and young adults—State of the art. Saudi J. Gastroenterol. 2022, 28, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Bitter, R.M.; Oh, S.; Deng, Z.; Rahman, S.; Hite, R.K.; Yuan, P. Structure of the Wilson disease copper transporter ATP7B. Sci. Adv. 2022, 8, eabl5508. [Google Scholar] [CrossRef]
- Yang, G.M.; Xu, L.; Wang, R.M.; Tao, X.; Zheng, Z.W.; Chang, S.; Ma, D.; Zhao, C.; Dong, Y.; Wu, S.; et al. Structures of the human Wilson disease copper transporter ATP7B. Cell Rep. 2023, 42, 112417. [Google Scholar] [CrossRef]
- Mazi, T.A.; Shibata, N.M.; Medici, V. Lipid and energy metabolism in Wilson disease. Liver Res. 2020, 4, 5–14. [Google Scholar] [CrossRef]
- Rieber, M. Cancer Pro-oxidant therapy through copper redox cycling: Repurposing disulfiram and tetrathiomolybdate. Curr. Pharm. Des. 2020, 26, 4461–4466. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: Mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef] [PubMed]
- Schlaug, G.; Hefter, H.; Nebeling, B.; Engelbrecht, V.; Weiss, P.; Stöcklin, G.; Seitz, R.J. Dopamine D2 receptor binding and cerebral glucose metabolism recover after D-penicillamine-therapy in Wilson’s disease. J. Neurol. 1994, 241, 577–584. [Google Scholar] [CrossRef]
- Huang, C.C.; Chu, N.S.; Yen, T.C.; Wai, Y.Y.; Lu, C.S. Dopamine transporter binding in Wilson’s disease. Can. J. Neurol. Sci. 2003, 30, 163–167. [Google Scholar] [CrossRef]
- Kalita, J.; Tripathi, A.; Jadhav, M.; Thakur, R.S.; Patel, D.K. A study of dopaminergic pathway in neurologic Wilson disease with movement disorder. Mol. Neurobiol. 2023, 60, 3496–3506. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Antos, A.; Sorysz, Z.; Litwin, T. Psychiatric symptoms in Wilson’s disease-Consequence of ATP7B gene mutations or just coincidence?-Possible causal cascades and molecular pathways. Int. J. Mol. Sci. 2024, 25, 12354. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, J.C. Kayser-Fleischer ring. J. Postgrad. Med. 2008, 54, 238–240. [Google Scholar] [CrossRef]
- Stättermayer, A.F.; Entenmann, A.; Gschwantler, M.; Zoller, H.; Hofer, H.; Ferenci, P. The dilemma to diagnose Wilson disease by genetic testing alone. Eur. J. Clin. Investig. 2019, 49, e13147. [Google Scholar] [CrossRef] [PubMed]
- Denny-Brown, D.; Porter, H. The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson’s disease). N. Engl. J. Med. 1951, 245, 917–925. [Google Scholar] [CrossRef]
- Vilensky, J.A.; Redman, K. British anti-Lewisite (dimercaprol): An amazing history. Ann. Emerg. Med. 2003, 41, 378–383. [Google Scholar] [CrossRef]
- Walshe, J.M. Wilson’s disease; new oral therapy. Lancet 1956, 270, 25–26. [Google Scholar] [CrossRef]
- Walshe, J.M. Treatment of Wilson’s disease with trientine (triethylene tetramine) dihydrochloride. Lancet 1982, 1, 643–647. [Google Scholar] [CrossRef]
- Pfeiffenberger, J.; Kruse, C.; Mutch, P.; Harker, A.; Weiss, K.H. The steady state pharmacokinetics of trientine in Wilson disease patients. Eur. J. Clin. Pharmacol. 2018, 74, 731–736. [Google Scholar] [CrossRef]
- Humphries, W.R.; Mills, C.F.; Greig, A.; Roberts, L.; Inglis, D.; Halliday, G.J. Use of ammonium tetrathiomolybdate in the treatment of copper poisoning in sheep. Vet. Rec. 1986, 119, 596–598. [Google Scholar] [PubMed]
- Humphries, W.R.; Morrice, P.C.; Bremner, I. A convenient method for the treatment of chronic copper poisoning in sheep using subcutaneous ammonium tetrathiomolybdate. Vet. Rec. 1988, 123, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Dick, R.D.; Yuzbasiyan-Gurkin, V.; Tankanow, R.; Young, A.B.; Kluin, K.J. Initial therapy of patients with Wilson’s disease with tetrathiomolybdate. Arch. Neurol. 1991, 48, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Moura, J.J.G. Diverse biological roles of the tetrathiomolybdate anion. Coord. Chem. Rev. 2021, 429, 213635. [Google Scholar] [CrossRef]
- Weiss, K.H.; Askari, F.K.; Czlonkowska, A.; Ferenci, P.; Bronstein, J.M.; Bega, D.; Ala, A.; Nicholl, D.; Flint, S.; Olsson, L.; et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: An open-label, multicentre, phase 2 study. Lancet Gastroenterol. Hepatol. 2017, 2, 869–876. [Google Scholar] [CrossRef]
- Froitzheim, G.; Olligs, H. Tierexperimentelle Untersuchungen zur Behandlung der Quecksilbervergiftung mit alpha,alpha 1-Dimercaptobernsteinsäure [Experimental studies on the treatment of mercurial poisoning with alpha,alpha 1-dimercaptosuccinic acid]. Dermatol. Wochenschr. 1954, 129, 497–500. [Google Scholar] [PubMed]
- Graziano, J.H.; Cuccia, D.; Friedheim, E. The pharmacology of 2,3-dimercaptosuccinic acid and its potential use in arsenic poisoning. J. Pharmacol. Exp. Ther. 1978, 207, 1051–1055. [Google Scholar] [CrossRef]
- Graziano, J.H.; Leong, J.K.; Friedheim, E. 2,3-Dimercaptosuccinic acid: A new agent for the treatment of lead poisoning. J. Pharmacol. Exp. Ther. 1978, 206, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.S.; Liang, Y.Y. Antidotal effects of dimercaptosuccinic acid. J. Appl. Toxicol. 1991, 11, 7–14. [Google Scholar] [CrossRef]
- Yang, R. Clinical analysis of 418 patients with Wilson’s disease in traditional Chinese medicine and Western medicine therapy. Chin. J. Mod. Dev. Tradit. Med. 1990, 10, 134–136. (In Chinese) [Google Scholar] [PubMed]
- Aposhian, H.V. DMSA and DMPS--water soluble antidotes for heavy metal poisoning. Annu. Rev. Pharmacol. Toxicol. 1983, 23, 193–215. [Google Scholar] [CrossRef]
- Zhu, X.Q.; Li, L.Y.; Yang, W.M.; Wang, Y. Combined dimercaptosuccinic acid and zinc treatment in neurological Wilson’s disease patients with penicillamine-induced allergy or early neurological deterioration. Biosci Rep. 2020, 40, BSR20200654. [Google Scholar] [CrossRef]
- Paolieri, M. Ferdinant Münz: EDTA and 40 years of inventions. Bull. Hist. Chem. 2017, 42, 133–140. [Google Scholar] [CrossRef]
- Stamelou, M.; Tuschl, K.; Chong, W.K.; Burroughs, A.K.; Mills, P.B.; Bhatia, K.P.; Clayton, P.T. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: A new treatable disorder. Mov. Disord. 2012, 27, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef]
- Lamas, G.A.; Issa, O.M. Edetate disodium-based treatment for secondary prevention in post-myocardial infarction patients. Curr. Cardiol. Rep. 2016, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Lamas, G.A.; Goertz, C.; Boineau, R.; Mark, D.B.; Rozema, T.; Nahin, R.L.; Lindblad, L.; Lewis, E.F.; Drisko, J.; Lee, K.L.; et al. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: The TACT randomized trial. JAMA 2013, 309, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Kirk, F.T.; Munk, D.E.; Swenson, E.S.; Quicquaro, A.M.; Vendelbo, M.H.; Larsen, A.; Schilsky, M.L.; Ott, P.; Sandahl, T.D. Effects of tetrathiomolybdate on copper metabolism in healthy volunteers and in patients with Wilson disease. J. Hepatol. 2024, 80, 586–595. [Google Scholar] [CrossRef]
- Schouwink, C. De Hepato-Cerebrale Degeneratie (Met Een Onderzoek van de Zinktofwisseling (Hepatolenticular Degeneration: Treatment by Oral Zinc). Master’s Thesis, University of Amsterdam, Amsterdam, The Netherlands, 1961. [Google Scholar]
- Lee, D.Y.; Brewer, G.J.; Wang, Y.X. Treatment of Wilson’s disease with zinc. VII. Protection of the liver from copper toxicity by zinc-induced metallothionein in a rat model. J. Lab. Clin. Med. 1989, 114, 639–645. [Google Scholar] [PubMed]
- Appenzeller-Herzog, C.; Mathes, T.; Heeres, M.L.S.; Weiss, K.H.; Houwen, R.H.J.; Ewald, H. Comparative effectiveness of common therapies for Wilson disease: A systematic review and meta-analysis of controlled studies. Liver Int. 2019, 39, 2136–2152. [Google Scholar] [CrossRef]
- Chen, J.C.; Chuang, C.H.; Wang, J.D.; Wang, C.W. Combination therapy using chelating agent and zinc for Wilson’s disease. J. Med. Biol. Eng. 2015, 35, 697–708. [Google Scholar] [CrossRef]
- Chen, D.; Zhou, X.; Hou, H.; Feng, L.; Liu, J.; Liang, Y.; Lin, X.; Zhang, J.; Wu, C.; Liang, X.; et al. Clinical efficacy of combined sodium dimercaptopropanesulfonate and zinc treatment in neurological Wilson’s disease with D-penicillamine treatment failure. Ther. Adv. Neurol. Disord. 2016, 9, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Jagadisan, B.; Dhawan, A. Combination treatment with chelators and zinc for Wilson disease: A double-edged sword. J. Clin. Exp. Hepatol. 2024, 14, 101372. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, H.; Inoue, I.; Inui, A.; Komatsu, H.; Sogo, T.; Murayama, K.; Murakami, T.; Yorifuji, T.; Asayama, K.; Katayama, S.; et al. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: Hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr. Res. 2006, 60, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A. Wilson Disease: Copper-mediated cuproptosis, iron-related ferroptosis, and clinical highlights, with comprehensive and critical analysis update. Int. J. Mol. Sci. 2024, 25, 4753. [Google Scholar] [CrossRef]
- Weiskirchen, R. Hepatoprotective and Anti-fibrotic agents: It’s time to take the next step. Front. Pharmacol. 2016, 6, 303. [Google Scholar] [CrossRef]
- Song, D.; Takahashi, G.; Zheng, Y.W.; Matsuo-Takasaki, M.; Li, J.; Takami, M.; An, Y.; Hemmi, Y.; Miharada, N.; Fujioka, T.; et al. Retinoids rescue ceruloplasmin secretion and alleviate oxidative stress in Wilson’s disease-specific hepatocytes. Hum. Mol. Genet. 2022, 31, 3652–3671. [Google Scholar] [CrossRef]
- Gromadzka, G.; Czerwińska, J.; Krzemińska, E.; Przybyłkowski, A.; Litwin, T. Wilson’s disease-crossroads of genetics, inflammation and immunity/autoimmunity: Clinical and molecular issues. Int. J. Mol. Sci. 2024, 25, 9034. [Google Scholar] [CrossRef]
- National Library of Medicine. PubChem Compound. Available online: https://www.ncbi.nlm.nih.gov/pccompound/ (accessed on 12 June 2025).
- Hou, H.; Chen, D.; Liu, J.; Feng, L.; Zhang, J.; Liang, X.; Xu, Y.; Li, X. Clinical and genetic analysis in neurological Wilson’s disease patients with neurological worsening following chelator therapy. Front. Genet. 2022, 13, 875694. [Google Scholar] [CrossRef]
- Gauthier, L.; Charbonnier, P.; Chevallet, M.; Delangle, P.; Texier, I.; Gateau, C.; Deniaud, A. Development, formulation, and cellular mechanism of a lipophilic copper chelator for the treatment of Wilson’s disease. Int. J. Pharm. 2022, 609, 121193. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Sharma, S.; Shukla, R. Nano-mediated molecular targeting in diagnosis and mitigation of Wilson disease. Mol. Neurobiol. 2024, 61, 4240–4258. [Google Scholar] [CrossRef]
- Perera, V.S.; Liu, H.; Wang, Z.Q.; Huang, S.D. Cell Permeable Au@ZnMoS4 core-shell nanoparticles: Towards a novel cellular copper detoxifying drug for Wilson’s disease. Chem. Mater. 2013, 25, 4703–4709. [Google Scholar] [CrossRef]
- Kandanapitiye, M.S.; Gunathilake, C.; Jaroniec, M.; Huang, S.D. Biocompatible D-Penicillamine conjugated Au nanoparticles: Targeting intracellular free copper ions for cetoxification. J. Mater. Chem. B. 2015, 3, 5553–5559. [Google Scholar] [CrossRef] [PubMed]
- Francis, C.; Wroblewska, L.; Pegman, P.; Amiji, M. Systemic biodistribution and hepatocyte-specific gene editing with CRISPR/Cas9 using hyaluronic acid-based nanoparticles. Nanomedicine 2022, 40, 102488. [Google Scholar] [CrossRef]
- Eker, F.; Duman, H.; Akdaşçi, E.; Bolat, E.; Sarıtaş, S.; Karav, S.; Witkowska, A.M. A Comprehensive review of nanoparticles: From classification to application and toxicity. Molecules 2024, 29, 3482. [Google Scholar] [CrossRef] [PubMed]
- Tremmel, R.; Uhl, P.; Helm, F.; Wupperfeld, D.; Sauter, M.; Mier, W.; Stremmel, W.; Hofhaus, G.; Fricker, G. Delivery of Copper-chelating Trientine (TETA) to the central nervous system by surface modified liposomes. Int. J. Pharm. 2016, 512, 87–95. [Google Scholar] [CrossRef]
- Padula, A.; Spinelli, M.; Nusco, E.; Bujanda Cundin, X.; Capolongo, F.; Campione, S.; Perna, C.; Bastille, A.; Ericson, M.; Wang, C.C.; et al. Genome editing without nucleases confers proliferative advantage to edited hepatocytes and corrects Wilson disease. JCI Insight 2023, 8, e171281. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Felice, C.; Rivera, K.; Pappin, D.J.; Tonks, N.K. DPM-1001 decreased copper levels and ameliorated deficits in a mouse model of Wilson’s disease. Genes. Dev. 2018, 32, 944–952. [Google Scholar] [CrossRef]
- Monestier, M.; Pujol, A.M.; Lamboux, A.; Cuillel, M.; Pignot-Paintrand, I.; Cassio, D.; Charbonnier, P.; Um, K.; Harel, A.; Bohic, S.; et al. A liver-targeting Cu(i) chelator relocates Cu in hepatocytes and promotes Cu excretion in a murine model of Wilson’s disease. Metallomics 2020, 12, 1000–1008. [Google Scholar] [CrossRef]
- Esmieu, C.; Hostachy, S.; Hureau, C. Cu(I) chelators: Useful tools to reveal and control Cu(I) homeostasis and toxicity. Coord. Chem. Rev. 2025, 539, 216684. [Google Scholar] [CrossRef]
- Vetrik, M.; Mattova, J.; Mackova, H.; Kucka, J.; Pouckova, P.; Kukackova, O.; Brus, J.; Eigner-Henke, S.; Sedlacek, O.; Sefc, L.; et al. Biopolymer strategy for the treatment of Wilson’s disease. J. Control. Release 2018, 273, 131–138. [Google Scholar] [CrossRef]
- Lichtmannegger, J.; Leitzinger, C.; Wimmer, R.; Schmitt, S.; Schulz, S.; Kabiri, Y.; Eberhagen, C.; Rieder, T.; Janik, D.; Neff, F.; et al. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J. Clin. Investig. 2016, 126, 2721–2735. [Google Scholar] [CrossRef] [PubMed]
- Berzina, A.; Martinsone, I.; Svirskis, S.; Murovska, M.; Kalis, M. Curcumin effect on copper transport in HepG2 cells. Medicina 2018, 54, 14. [Google Scholar] [CrossRef]
- van den Berghe, P.V.; Stapelbroek, J.M.; Krieger, E.; de Bie, P.; van de Graaf, S.F.; de Groot, R.E.; van Beurden, E.; Spijker, E.; Houwen, R.H.; Berger, R.; et al. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology 2009, 50, 1783–1795. [Google Scholar] [CrossRef]
- Spincemaille, P.; Pham, D.H.; Chandhok, G.; Verbeek, J.; Zibert, A.; Libbrecht, L.; Schmidt, H.; Esguerra, C.V.; de Witte, P.A.; Cammue, B.P.; et al. The plant decapeptide OSIP108 prevents copper-induced toxicity in various models for Wilson disease. Toxicol. Appl. Pharmacol. 2014, 280, 345–351. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Koganti, L.; Muchenditsi, A.; Pendyala, V.S.; Huso, D.; Hankin, J.; Murphy, R.C.; Huster, D.; Merle, U.; Mangels, C.; et al. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B−/− (Wilson disease) mice. Hepatology 2016, 63, 1828–1841. [Google Scholar] [CrossRef] [PubMed]
- Pujol, A.M.; Cuillel, M.; Renaudet, O.; Lebrun, C.; Charbonnier, P.; Cassio, D.; Gateau, C.; Dumy, P.; Mintz, E.; Delangle, P. Hepatocyte targeting and intracellular copper chelation by a thiol-containing glycocyclopeptide. J. Am. Chem. Soc. 2011, 133, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Petruzzelli, R.; Catalano, F.; Crispino, R.; Polishchuk, E.V.; Elia, M.; Masone, A.; Lavigna, G.; Grasso, A.; Battipaglia, M.; Sepe, L.V.; et al. Prion protein promotes copper toxicity in Wilson disease. Nat. Commun. 2025, 16, 1468. [Google Scholar] [CrossRef]
- van Meer, S.; de Man, R.A.; van den Berg, A.P.; Houwen, R.H.; Linn, F.H.; van Oijen, M.G.; Siersema, P.D.; van Erpecum, K.J. No increased risk of hepatocellular carcinoma in cirrhosis due to Wilson disease during long-term follow-up. J. Gastroenterol. Hepatol. 2015, 30, 535–539. [Google Scholar] [CrossRef]
- Shribman, S.; Bocchetta, M.; Sudre, C.H.; Acosta-Cabronero, J.; Burrows, M.; Cook, P.; Thomas, D.L.; Gillett, G.T.; Tsochatzis, E.A.; Bandmann, O.; et al. Neuroimaging correlates of brain injury in Wilson’s disease: A multimodal, whole-brain MRI study. Brain 2022, 145, 263–275. [Google Scholar] [CrossRef]
- Immergluck, J.; Anilkumar, A.C. Wilson Disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441990/ (accessed on 7 August 2023).
- Cai, S.; Gong, J.Y.; Yang, J.; Wang, J.S. Anemia following zinc treatment for Wilson’s disease: A case report and literature review. BMC Gastroenterol. 2019, 19, 120. [Google Scholar] [CrossRef]
- Chevalier, K.; Obadia, M.A.; Djebrani-Oussedik, N.; Poujois, A. Can patients with Wilson’s disease develop copper deficiency? Mov. Disord. Clin. Pract. 2023, 10, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Tornabene, D.; Bini, P.; Gastaldi, M.; Vegezzi, E.; Asteggiano, C.; Marchioni, E.; Diamanti, L. Neurological complications due to copper deficiency in the context of Wilson disease treatment: A case report with long-term follow-up and review of the literature. Neurol. Sci. 2024, 45, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Li, W.; Jiang, H.; Yin, Z.; Zhou, Z. Characterization of nerve biopsy in copper deficiency peripheral neuropathy due to over-treatment of Wilson’s disease: A case report. Acta Neurol. Belg. 2025, 125, 579–582. [Google Scholar] [CrossRef]
- Weiskirchen, S.; Weiskirchen, R. Unraveling the future: Hot topics shaping molecular diagnostics today. Expert. Rev. Mol. Diagn. 2025, 25, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L. Current and emerging issues in Wilson’s disease. N. Engl. J. Med. 2023, 389, 922–938. [Google Scholar] [CrossRef]
- Vidhusree, D.; Ap, K. Wilson’s disease in childhood and the challenges in its diagnosis: A case report. Cureus 2024, 16, e65847. [Google Scholar] [CrossRef]
- Selimoglu, M.A.; Ertekin, V.; Doneray, H.; Yildirim, M. Bone mineral density of children with Wilson disease: Efficacy of penicillamine and zinc therapy. J. Clin. Gastroenterol. 2008, 42, 194–198. [Google Scholar] [CrossRef]
- Sharma, N.; Das, D.D.C. Exploring the potential of trientine tetrahydrochloride in the treatment of Wilson disease. Health Sci. Rev. 2023, 6, 100082. [Google Scholar] [CrossRef]
- Ghosh, U.; Sen Sarma, M.; Samanta, A. Challenges and dilemmas in pediatric hepatic Wilson’s disease. World J. Hepatol. 2023, 15, 1109–1126. [Google Scholar] [CrossRef]
- Ehsan Arshad, H.M.; Zain Raza, M.; Maqsood, M.; Omais, M.; Faisal, M.H.; Nadeem, A.A. Comparative analysis of clinical outcomes and safety profile of trientine and d-penicillamine in the management of Wilson’s disease: A systematic review and meta-analysis. Rare 2025, 3, 100077. [Google Scholar] [CrossRef]
- Boaru, S.G.; Merle, U.; Uerlings, R.; Zimmermann, A.; Weiskirchen, S.; Matusch, A.; Stremmel, W.; Weiskirchen, R. Simultaneous monitoring of cerebral metal accumulation in an experimental model of Wilson’s disease by laser ablation inductively coupled plasma mass spectrometry. BMC Neurosci. 2014, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, T.D.; Medici, V. Edging closer to successful gene therapy for Wilson disease. Mol. Ther. Methods Clin. Dev. 2022, 27, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, G.A.; Fang, S.; Li, X.; Ding, Y.; Song, Y.; He, W.; Rao, Z.; Diao, K.; Zhu, X.; et al. Decoding Wilson disease: A machine learning approach to predict neurological symptoms. Front. Neurol. 2024, 15, 1418474. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Name | 2,3-Dimercapto-1-propanol (Dimercaprol, Dithioglycerin), commonly referred to as British anti-lewisite (BAL) |
| CAS No. | 59-52-9 |
| Chemical Formula | C3H8O2S2 |
| Structure | 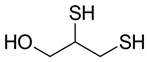 |
| Basic Description | A colorless or slightly yellow liquid with two sulfhydryl (–SH) groups, originally developed as an antidote to the chemical warfare agent lewisite. |
| Mechanism of Action | Forms stable, water-soluble complexes with heavy metals such as arsenic, gold, lead, and mercury. Although not a primary treatment for Wilson’s disease, BAL can chelate copper in acute settings. |
| Solubility | In water, BAL is slightly soluble and often unstable in aqueous solutions. However, in organic solvents or oils, it is more soluble, and typically formulated in peanut oil for intramuscular injection. |
| Therapeutic Use in Wilson’s Disease | The primary indication for BAL is an antidote for acute heavy metal poisoning (e.g., arsenic, gold, mercury, lead). In cases of Wilson’s disease, BAL has historically been considered in acute or special scenarios, but is rarely used today due to significant side effects and the availability of better chelators (e.g., penicillamine, trientine). The dosage range for BAL is commonly between 2.5 and 5 mg/kg via intramuscular injection, with frequency guided by clinical protocols and the severity of poisoning. |
| Adverse Effects and Monitoring | Common side effects include hypertension, tachycardia, gastrointestinal distress, headache, and local injection site reactions. Monitoring of vital signs, complete blood count, and serum metal levels (where applicable) is necessary to prevent under- or overtreatment and detect toxicity. |
| Other relevant information | The clinical use of BAL has declined in favor of newer agents with fewer adverse effects. Treatment with BAL requires careful medical supervision due to its potential toxicity and the complexity of managing acute heavy metal exposures. |
| Chemical Name | D-Penicillamine (3-Mercapto-D-valine; 3,3-Dimethyl-D-cysteine) |
| CAS No. | 52-67-5 |
| Chemical Formula | C5H11NO2S |
| Structure |  |
| Basic Description | A sulfur-containing amino acid derivative historically related to penicillin, though lacking antibiotic properties. |
| Mechanism of Action | It primarily functions as a copper-chelating agent, binding free copper ions and promoting their excretion in urine. It also has some chelating activity for other metals (e.g., lead, mercury) and is occasionally used for rheumatoid arthritis. |
| Solubility | Penicillamine is moderately to freely soluble in water (depending on pH) and sparingly soluble in ethanol. In an aqueous alkaline solution (e.g., 1N sodium hydroxide), it is readily soluble. |
| Therapeutic Use in Wilson’s Disease | The typical dosage range for adults is 750–1500 mg per day, divided into two or three doses and given orally as capsules or tablets. It should be administered on an empty stomach, at least 1 h before meals and 2 h after meals. Concentration in therapeutic use varies among individuals, but dosing is generally titrated based on clinical response and copper-level monitoring. |
| Adverse Effects and Monitoring | Common side effects include gastrointestinal discomfort, skin rash, bone marrow suppression (e.g., thrombocytopenia, leukopenia), renal impairment, diarrhea, loss of appetite, and, in rare cases, a lupus-like syndrome. Regular blood counts, liver function tests, and 24-h urinary copper levels are essential for monitoring to balance effective copper chelation with the risk of toxicity. |
| Other relevant information | Initiation of therapy may temporarily worsen neurologic symptoms in some patients with Wilson’s disease; careful initiation and monitoring are recommended. Patients often require lifelong therapy to maintain adequate copper control and prevent accumulation. Adequate dietary measures (e.g., reduced copper intake) may complement medical treatment. Penicillamine is sold under the trade name Cuprimine and used in the therapy of cystinuria, rheumatoid arthritis, scleroderma, and lead poisoning if no other preferred chelating agents are available. However, it is contraindicated in patients with a previous history of penicillamine-related aplastic anemia, penicillin allergy, or renal insufficiency. |
| Chemical Name | Triethylenetetramine dihydrochloride (commonly referred to as Trientine, TETA) |
| CAS No. | 38260-01-4 (Dihydrochloride), 4961-40-4 (Tetrahydrochloride) |
| Chemical Formula | C6H18N4 × 2HCl (for the dihydrochloride salt) |
| Structure | 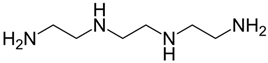 |
| Basic Description | A polyamine compound designed to chelate copper ions and aid in their excretion. It shares a similar therapeutic indication to penicillamine but is structurally distinct. |
| Mechanism of Action | It binds copper ions in the bloodstream and tissues, forming complexes that are excreted via the urine. It is particularly useful for patients with Wilson’s disease who experience intolerance or adverse effects with penicillamine. |
| Solubility | Trientine is freely soluble in water, while having limited solubility in common organic solvents. |
| Therapeutic Use in Wilson’s Disease | The typical dosage range is 750–1500 mg per day (divided doses) for adults, though some regimens may go up to 2 g per day depending on clinical requirements. The drug is usually administered in an oral formulation (capsule) for convenience and consistent dosing. The goal is to maintain a therapeutic schedule that effectively lowers copper load while minimizing adverse effects. |
| Adverse Effects and Monitoring | Common side effects include gastrointestinal discomfort, iron deficiency anemia (due to chelation of iron), and possible hypersensitivity reactions. |
| In therapy monitoring, regular measures of 24-h urinary copper excretion and serum free copper are suggested to assess treatment effectiveness, along with periodic blood counts to check for anemia and other cytopenias. | |
| Other relevant information | It is typically regarded as a second-line option for patients who cannot tolerate penicillamine or require alternative chelation therapy. Long-term therapy is often necessary to keep copper levels controlled, with patient compliance and close medical follow-up being paramount. Dietary counseling to limit excessive copper intake (e.g., avoiding copper-rich foods) can further support therapeutic goals. Trientine tetrahydrochloride has been sold under the brand name Cuprior in Europe since 2019 and under the brand name Cuvrior in the US since 2022. Trientine has been found to reduce serum iron levels, and iron supplements may be necessary in case of iron deficiency anemia. The combination of trientine with zinc is not recommended. |
| Chemical Name | Tetrathiomolybdate (typically used in the form of ammonium tetrathiomolybdate, TTM) |
| CAS No. | 15060-55-6 (Ammonium tetrathiomolybdate) |
| Chemical Formula | (NH4)2MoS4 |
| Structure | 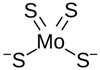 (Molybdate) |
| Basic Description | A molybdenum-sulfide compound that forms strong complexes with copper, reducing copper availability in the body. |
| Mechanism of Action | It binds free copper in the blood and tissues, promoting the formation of excretable complexes and reducing intestinal copper absorption by forming insoluble copper complexes in the gastrointestinal tract. |
| Solubility | In water, it is soluble, with solubility varying depending on pH and salt form. In organic solvents, it generally has poor solubility. |
| Therapeutic Use in Wilson’s Disease | The typical dosage is often initiated in research or compassionate-use protocols with regimens varying but ranging from about 60 to 120 mg/day in divided doses, depending on disease severity and patient response. It is particularly considered for patients with neurological involvement or those intolerant to other chelators. Sometimes used in short-term “de-coppering” protocols, long-term approaches exist in specific clinical trial settings. |
| Adverse Effects and Monitoring | Potential side effects include overcorrection leading to copper deficiency (e.g., anemia, neutropenia), gastrointestinal disturbances, and possible bone marrow suppression. Close monitoring of hematological parameters, serum copper levels, and clinical response is crucial to prevent inadequate or excessive copper depletion. |
| Other relevant information | Availability varies and can be limited outside of research settings. Continued investigation aims to clarify optimal dosing, duration of therapy, and long-term safety. Patient compliance and a multidisciplinary care plan, including dietary counseling, play critical roles in successful management. TTM for oral application in Wilson’s disease is sold under the brand name Coprexa. Dihydrogentetramolybdate (tiomobibdic acid) is sold under the trade name Decuprate. The bis-choline complexed form of tetramolybdate, also known as WTX-101 or ALXN1840, is an effective drug that targets hepatic intracellular copper and reduces nonceruloplasmin-bound copper by forming tripartite complexes with albumin and increasing biliary copper excretion [45]. |
| Chemical Name | 2,3-Dimercaptosuccinic acid (Meso-2,3-dimercaptosuccinic acid), commonly referred to as DMSA, sometimes called Dimercaptosuccinate in certain contexts |
| CAS No. | 304-55-2 |
| Chemical Formula | C4H6O4S2 |
| Structure | 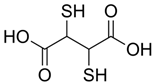 |
| Basic Description | This water-soluble dithiol-containing chelator with two sulfhydryl (–SH) groups is capable of binding heavy metals such as lead, mercury, and arsenic. Commercially available forms include the disodium salt (more soluble) and the free acid. |
| Mechanism of Action | It chelates metal ions by forming stable, water-soluble complexes that are primarily excreted via the kidneys. Widely recognized for its use in managing lead poisoning, it also shows efficacy against other heavy metals. |
| Solubility | The free acid form is sparingly soluble in water, while salt forms (e.g., disodium salt) have better solubility in water, which can be advantageous for oral dosing. |
| Therapeutic Use in Wilson’s Disease | Its primary indication is the oral treatment of children and adults with elevated blood lead levels (succimer). In addition to Pb2+, it forms complexes with several other divalent cations, including Cd2+, Fe2+, Hg2+, Zn2+, and Ni2+. It also shows chelating activity against mercury and arsenic. While not a standard agent for Wilson’s disease, it is commonly managed with penicillamine, trientine, or zinc. The typical dosage for lead poisoning is 10 mg/kg orally every eight hours for five days, with specific protocols varying by region and clinical practice. |
| Adverse Effects and Monitoring | Common side effects include gastrointestinal discomfort, mild skin rash, and transient elevated liver enzymes in some cases.Monitoring should include periodic blood counts, renal function tests, and metal levels (e.g., lead) to ensure effective chelation and avoid overcorrection. |
| Other relevant information | It is safe and effective for outpatient management of moderate lead toxicity, especially in children. It is less lipid-soluble than some older chelators (e.g., Dimercaprol/BAL), resulting in fewer central nervous system side effects. Compliance and adherence to dosing schedules are crucial for successful chelation therapy, often combined with environmental controls (e.g., removing the source of lead). It is absorbed rapidly but incompletely after oral administration. DMSA in plasma is mainly albumin-bound, with only a very small amount present as a free drug. |
| Chemical Name | Ethylenediaminetetraacetic acid (EDTA) |
| CAS No. | 60-00-4 |
| Chemical Formula | C10H16N2O8 |
| Structure | 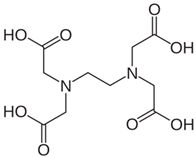 |
| Basic Description | EDTA is a chelating agent with four carboxyl groups and two amine groups, forming stable complexes with many metal ions. |
| Mechanism of Action | It binds divalent and trivalent metal ions (e.g., Pb2+, Ca2+, Fe3+) to form water-soluble complexes that are excreted, primarily via the kidneys. It is used clinically in the form of calcium disodium EDTA (CaNa2EDTA) when treating certain heavy metal toxicities, such as lead poisoning. |
| Solubility | In water, EDTA is moderately soluble, and its solubility is enhanced by converting it to a sodium or calcium disodium salt.In other solvents, it is generally sparingly soluble in many organic solvents. |
| Therapeutic Use in Wilson’s Disease | A common indication for EDTA is the management of lead poisoning, often given alongside dimercaprol in severe cases. It is typically administered intravenously or intramuscularly in a hospital setting. The dosage for lead poisoning in adults is often 1000 mg/m2 per day (or around 30 mg/kg/day) as a continuous intravenous infusion or in divided doses. Exact regimens vary by age, body weight, and clinical protocols. |
| Adverse Effects and Monitoring | Potential side effects of EDTA include nephrotoxicity (especially at higher doses or prolonged administration), electrolyte disturbances (e.g., hypocalcemia), and local injection site irritation. Monitoring kidney function, electrolytes, and blood lead levels is routinely conducted to assess therapeutic efficacy and minimize the risk of toxicity. |
| Other relevant information | EDTA is utilized in laboratory settings as an anticoagulant in blood collection tubes and in specific industrial applications due to its potent metal-chelating properties. In medical contexts, it is crucial to differentiate between calcium disodium EDTA (used for lead chelation) and disodium EDTA (which can chelate calcium more aggressively, posing a greater risk of hypocalcemia if used incorrectly). Treatment for heavy metal poisoning is tailored to the particular metal, the clinical status of the patient, and any coexisting medical issues, often necessitating close consultation with toxicology specialists. Intravenous chelation therapy with disodium EDTA reduces the likelihood of adverse cardiovascular outcomes, such as atherosclerosis, by scavenging calcium found in fatty, atherosclerotic deposits [56,57]. EDTA is also known as edetic acid, Titriplex II, Trilon B, Idranal II, Edathamil (EDTA tetrasodium dehydrate), Chelaplex III (EDTA disodium salt), and Versene (EDTA solution in phosphate-buffered saline). |
| Chemical Name | Bis-choline tetrathiomolybdate (commonly referred to as WTX101). Also known under synonym ATN-224; bis-choline-TTM) |
| CAS No. | 649749-10-0 |
| Chemical Formula | C10H30N2O8 |
| Structure | 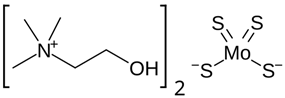 |
| Basic Description | A new formulation of TTM bound to choline moieties designed to improve oral bioavailability and tolerability compared to previous versions of TTM. |
| Mechanism of Action | It binds free copper ions in the bloodstream and gastrointestinal tract, forming complexes that reduce copper absorption and facilitate excretion, keeping “free” or nonceruloplasmin-bound copper at safe levels, thus helping prevent the toxic effects of copper overload. |
| Solubility | Primarily designed for oral administration, this choline-based composition offers sufficient solubility in aqueous environments for gastrointestinal absorption. Specific solubility may vary depending on the formulation and pH. |
| Therapeutic Use in Wilson’s Disease | This investigation agent is mainly being studied in clinical trials for the treatment of Wilson’s disease. Study protocols have examined different oral doses, typically in the tens of milligrams per day range, which are based on therapeutic response and copper-level monitoring. The drug is used to rapidly reduce and sustain low levels of exchangeable (free) copper without causing copper deficiency. |
| Adverse Effects and Monitoring | Possible side effects include the risk of inducing copper deficiency (e.g., anemia, leukopenia), gastrointestinal discomfort, and other mild to moderate events depending on the dosage. Close monitoring of free copper, total serum copper, ceruloplasmin, and blood counts is crucial. Patients should be evaluated for any hepatic or hematologic changes. |
| Other relevant information | Intended to offer a more convenient and potentially safer alternative to other copper-chelating regimens, although long-term effectiveness and safety are still being assessed. It is typically incorporated into a comprehensive management plan for Wilson’s disease, which may involve dietary recommendations to reduce copper intake. Availability may be limited to clinical trials or regulated programs until final regulatory approvals are obtained. Collaboration with toxicology specialists is often necessary. |
| Chemical Name | Zinc is an element, typically administered as zinc acetate or zinc sulfate in clinical settings |
| CAS No. | Zinc (element): CAS No. 7440-66-6; zinc acetate: CAS No. 557-34-6 (commonly used for Wilson’s disease therapy); zinc sulfate: CAS No. 7733-02-0 |
| Chemical Formula | Zn(CH3COO)2 (often formulated as zinc acetate dihydrate in capsules or tablets) |
| Structure | Zn2+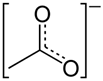 Zn2+ Zn2+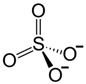 Zinc acetate (water free) Zinc sulfate (water free) |
| Basic Description | Zinc, when administered as a salt, is an oral supplement used to block the absorption of copper. |
| Mechanism of Action | Induction of metallothionein: Zinc triggers the production of intestinal metallothionein, which specifically binds to copper, reducing its absorption and promoting its excretion through feces. |
| Solubility | Zinc acetate dissolves readily in water, with its solubility affected by formulation and pH. Zinc sulfate, also water-soluble, is commonly found in supplement form. |
| Therapeutic Use in Wilson’s Disease | Typically, a daily dose of 150 mg of elemental zinc is recommended (often taken as 50 mg three times a day), although dosing may vary depending on clinical response and local guidelines. Standard formulations are oral capsules or tablets that should be taken on an empty stomach for optimal effectiveness. |
| Adverse Effects and Monitoring | Possible side effects of zinc include gastrointestinal discomfort (nausea, stomach irritation), a metallic taste, and occasional headaches. |
| Other relevant information | Long-term treatment is often continued indefinitely to prevent copper re-accumulation. Zinc acetate is approved for treating Wilson’s disease under the brand names Galzin in the US and Wilzin in Europe. |
| Compound | Mechanism | Efficacy | Dosing | Side Effects | Monitoring | Clinical Indication |
|---|---|---|---|---|---|---|
| British anti-lewisite (BAL, Dimercaprol) | Chelates arsenic and various heavy metals (including Cu), forming stable complexes. | Historically used for WD, but it is much less effective and not recommended as a routine therapy today. | Typically administered intramuscularly. The exact regimens vary, and BAL is not used for long-term management of WD. | Injection site reactions, elevated blood pressure, possible neurological worsening, GI discomfort. | Blood pressure, neurological status, basic labs; if used, monitor Cu levels to assess response. | Blood pressure, neurological status, basic labs; if used, monitor Cu levels to assess response. |
| D-Penicillamine | Forms soluble complexes with Cu and aids its excretion in urine. | Widely considered a first-line agent, particularly effective in symptomatic patients with hepatic and/or neurological symptoms. | Commonly 750–1500 mg/day (20 mg/kg/day) in divided doses; dosage can be increased (up to ~2 g/day) in severe cases. | Hypersensitivity reactions, bone marrow suppression, lupus-like syndrome, renal impairment, connective tissue disorders; nausea, hair loss, diarrhea, risk of neurological deterioration upon initiation, and many others. | CBC, renal function, LFTs, 24-h urinary Cu excretion, and clinical status. | Primary therapy for WD, although some patients switch to alternatives (e.g., trientine) due to side effects. |
| Trientine | Chelates Cu by forming complexes excreted in urine; lacks some reactive sulfur groups found in penicillamine. | Considered equally effective to D-penicillamine for many patients and is often used if there is penicillamine intolerance. | Typically 750–2000 mg/day (20 mg/kg/day) in divided doses. The exact dose depends on the clinical response. | Generally fewer side effects than penicillamine, but can cause iron deficiency anemia, GI irritation, skin rash, arthralgia, or neurological worsening in rare cases. | CBC, iron parameters (ferritin), 24-h urinary Cu excretion, LFTs, clinical status. | CBC, iron parameters (ferritin), 24-h urinary Cu excretion, LFTs, clinical status. |
| Tetrathiomolybdate (TTM) | Binds Cu in the gut and bloodstream, reducing absorption and free Cu levels. | Especially useful in neurological WD, as it may reduce the risk of initial neurological worsening compared to other chelators. | Approximately 120 mg/day in divided doses with meals (regimens vary in research and clinical practice). | Risk of Cu deficiency (overtreatment), bone marrow suppression (anemia, neutropenia), and potential toxicity if dosed incorrectly. | Serum Cu indices (free Cu and ceruloplasmin), CBC, and clinical signs of Cu deficiency. | Investigational or second-line in some regions; can be first-line for neurological manifestations in specific protocols. |
| Dimercaptosuccinic Acid (DMSA, Succimer) | A water-soluble chelator that binds various heavy metals (commonly used for lead poisoning); also has Cu-chelating capabilities. | Not a standard first-line agent for WD and is occasionally used off-label or in investigational studies. | Often ~30 mg/kg/day in divided doses for heavy metal poisoning; protocols for WD are not well-established. | GI discomfort, rash, and potential elevation in liver enzymes. | CBC, LFTs, Cu indices, clinical response. | Primarily indicated for lead poisoning; role in WD is limited and not widely adopted. |
| Ethylenediaminetetraacetic Acid (EDTA) | Chelates multiple metals; extensively used for lead and some other heavy metal toxicities. | Not considered effective enough for routine WD treatment. | Typically administered parenterally (IV); there is no standardized regimen for WD. | Nephrotoxicity, electrolyte imbalances, and hypotension, if infused too rapidly. | Renal function, electrolytes, and vital signs. | Standard therapy for lead poisoning; not recommended for WD. |
| Bis-choline Tetrathiomolybdate | Similar to TTM; forms complexes with Cu and reduces absorption, lowering free Cu in serum. | Early studies suggest it may reduce the risk of neurological deterioration, potentially being as effective or superior to older chelators. | Trial protocols often use ~120 mg/day in divided doses; exact regimens are still being refined. | Cu deficiency, bone marrow suppression (cytopenias), GI disturbances. | Cu deficiency, bone marrow suppression (cytopenias), GI disturbances. | Investigational or emerging therapy for WD; focus on neurological protection. |
| Zinc | Induces metallothionein in enterocytes, which binds Cu and prevents its absorption. | Useful as maintenance therapy or in presymptomatic/mild cases; often combined or used after initial chelation in symptomatic patients. | Commonly, 50 mg of elemental zinc taken thrice daily on an empty stomach when body weight is over 50 kg; should be lowered to 25 mg when body weight is under 50 kg. | GI irritation, reduced iron absorption (leading to anemia if unmonitored). | Full clinical assessment, Cu indices (serum Cu/ceruloplasmin), iron status (ferritin), LFTs. | Maintenance or monotherapy in mild or presymptomatic WD, as well as patients intolerant to more potent chelators. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskirchen, R. Comprehensive Pharmacological Management of Wilson’s Disease: Mechanisms, Clinical Strategies, and Emerging Therapeutic Innovations. Sci 2025, 7, 94. https://doi.org/10.3390/sci7030094
Weiskirchen R. Comprehensive Pharmacological Management of Wilson’s Disease: Mechanisms, Clinical Strategies, and Emerging Therapeutic Innovations. Sci. 2025; 7(3):94. https://doi.org/10.3390/sci7030094
Chicago/Turabian StyleWeiskirchen, Ralf. 2025. "Comprehensive Pharmacological Management of Wilson’s Disease: Mechanisms, Clinical Strategies, and Emerging Therapeutic Innovations" Sci 7, no. 3: 94. https://doi.org/10.3390/sci7030094
APA StyleWeiskirchen, R. (2025). Comprehensive Pharmacological Management of Wilson’s Disease: Mechanisms, Clinical Strategies, and Emerging Therapeutic Innovations. Sci, 7(3), 94. https://doi.org/10.3390/sci7030094