1. Introduction
Conformational methods for characterizing peptides are of interest when determining their flexible nature. This flexibility results in many native-like conformations with differences in physical properties such as energies, dihedral angles, the orientation of side chains, increased structural activity levels, etc. Many studies have focused on analyzing the conformational variations in linear peptides. Apart from these peptides, another interesting class of peptides to be studied using conformational methods is the class of cyclic peptides [
1,
2,
3]. This peptide class differs from regular peptides; their cyclic nature conformationally restricts them. Although these peptides have limited flexibility due to the constraints placed upon them, they are still capable of displaying a variety of different side chain orientations as well as variations in their secondary structural motifs. Many studies have explored this form of peptide in solution and crystal form. Ramakrishnan et al., 1985, studied the variations among these peptides regarding symmetries and possible hydrogen bonding patterns [
4]. Earlier structures reported in this class were tripeptides. Tripeptides are strained systems and only have a few conformations. A crystal structure of cyclo trisarcosyl exemplifies this [
5]. This structure commonly features a cis geometry in its peptide linkage. In 1968, Venkatachalam’s study showed the formation of three-fold symmetric structures [
6]. Manjula, in 1979, showed not only a symmetric structure but also a non-symmetric structure with two cis and one trans linkage [
7]. The results of these studies conclusively demonstrated that a symmetrical molecular configuration featuring exclusively cis linkages possesses a lower energy state compared to a structure incorporating two cis linkages and a single trans linkage. The experimental evidence supporting this comes from Kessler and colleagues’ work in both the solid and solution states, revealing a symmetric conformation in solution and an asymmetric structure in the solid state. The flexibility of the cyclic peptide’s structure is evident, despite the reduced number of residues.
Researchers have extended their studies on these peptides to tetrapeptides and pentapeptides. With 12 atoms in their cyclic ring, tetrapeptides are less strained than tripeptides. Tetrapeptides have an all-trans conformation in their peptide units. In 1970, Groth provided experimental evidence of peptides showing a preference for obtaining an equal probability of cis and trans linkages [
8]. The theoretical proof of this type of peptide having all-trans and alternative cis trans linkages was presented by Ramakrishnan and Sarathy, 1968, and Manjula, Ramakrishnan, 1979, respectively [
7,
9].
The backbone geometry of the pentapeptide ring does not determine its lower-energy conformation; instead, this is governed by the specific hydrogen bonding interactions present within the structure. A 4-1 beta turn and a 3-1 gamma turn make up the motif and backbone bonding pattern for pentapeptides. Ramakrishnan and Narasinga Rao, in 1982, theoretically determined the various conformations of these cyclic peptides [
10]. The experimental crystal structures, as detailed in a 1982 report by Mauger et al., illustrated the presence of a beta turn stabilized by a 4-1 hydrogen bond, a key structural feature within the molecular ring. Subsequent studies by Manjula in 1979 demonstrated that altering the orientation of this peptide enhances its stability by forming an additional gamma turn [
8,
11].
Higher-order cyclic hexapeptides have been studied the most extensively, with both theoretical and experimental methods. Researchers have synthesized this type of peptide easily compared to other peptide classes. The most significant characteristic of this peptide is that its backbone adopts an ordered structure and has a well-defined backbone ordering in the trans conformation [
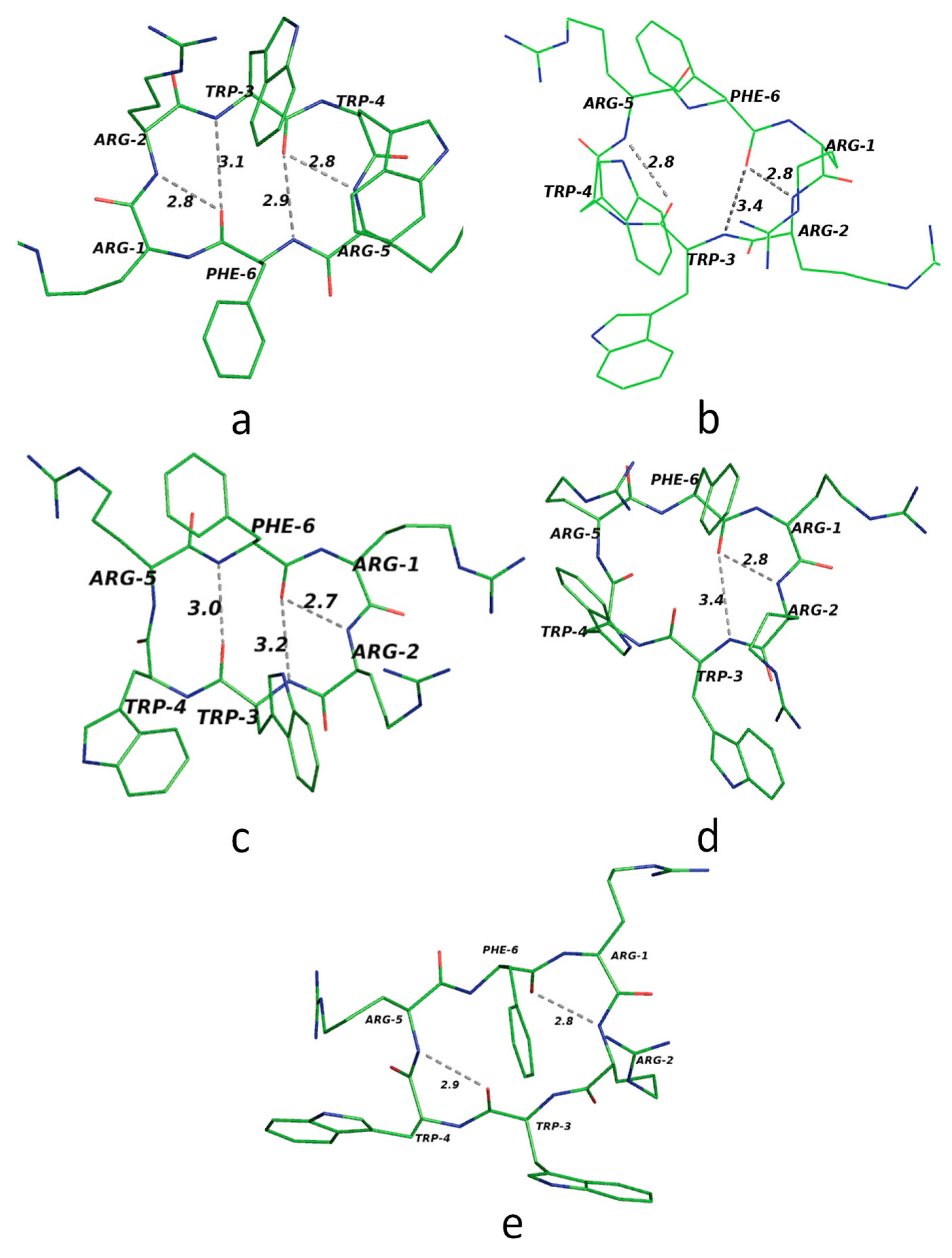
1]. Apart from this geometrical criterion, these peptides form structures with two beta turns. A type II beta turn shows a specific backbone orientation when the four Cα atoms of the corresponding residues lie in a single plane. If the structure has a beta type I turn, the backbone has a twist. Peptides with two type I beta turns have a perfect chair conformation, and they have a boat-like structure if their beta turn is type II. In nature, variations in this pattern exist, and peptides often have minor distortions. Several experimental results from detailed studies of sequence symmetry show that, in synthetic peptides, when all amino acid residues are of various types, they form two beta turns. Their corresponding hydrogen bonds stabilize these structures. An analysis of the crystal structure reveals that cyclic peptides with a tripeptide fragment share the same beta fold, resulting in two-fold symmetry. There is also the case of dissimilar tripeptide fragments being present. These peptides have two types of beta folds, one being the inverse fold of the other. The final case is that of two beta turns with completely dissimilar residues having different beta turns [
1].
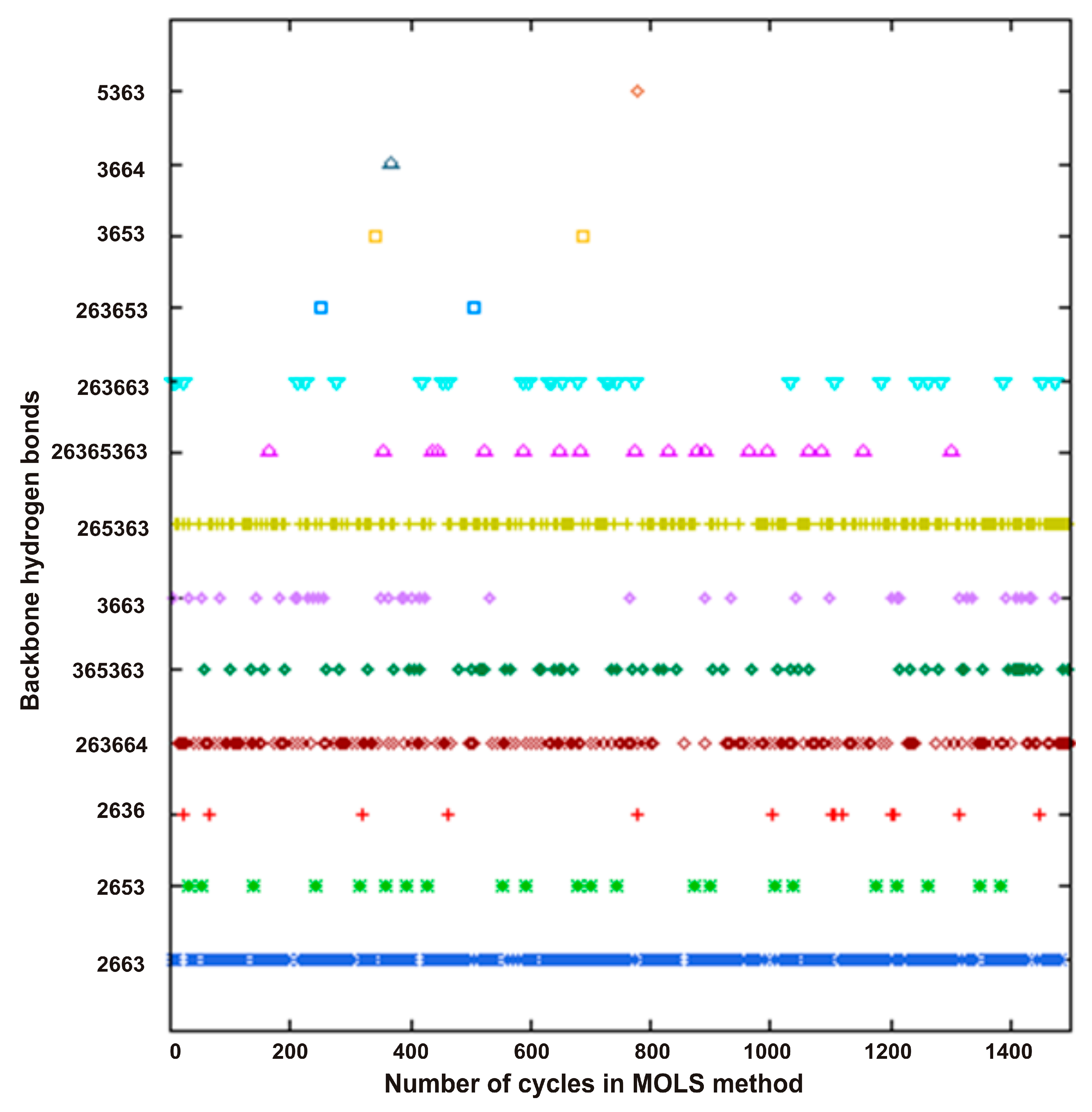
In their study, Ramakrishnan et al. [
10] found that, besides the above criteria, different hydrogen bonds stabilize peptides. They identified these bonds by utilizing stereochemical studies and energy calculations. Using the minimization technique on these peptides, they showed the presence of different hydrogen bonds with secondary structural motifs like beta turns and gamma turns. According to their results, there are various bonds that occur like the bifurcated bond between amino acid residues 4-1 and 3-1 with type I or the inverse type. Similarly, for the type II beta turn, the same acceptor oxygen forms hydrogen bonds. There are also parallel bonding patterns seen in between amino acid residues like 3N-6O and 6N-3O [
12].
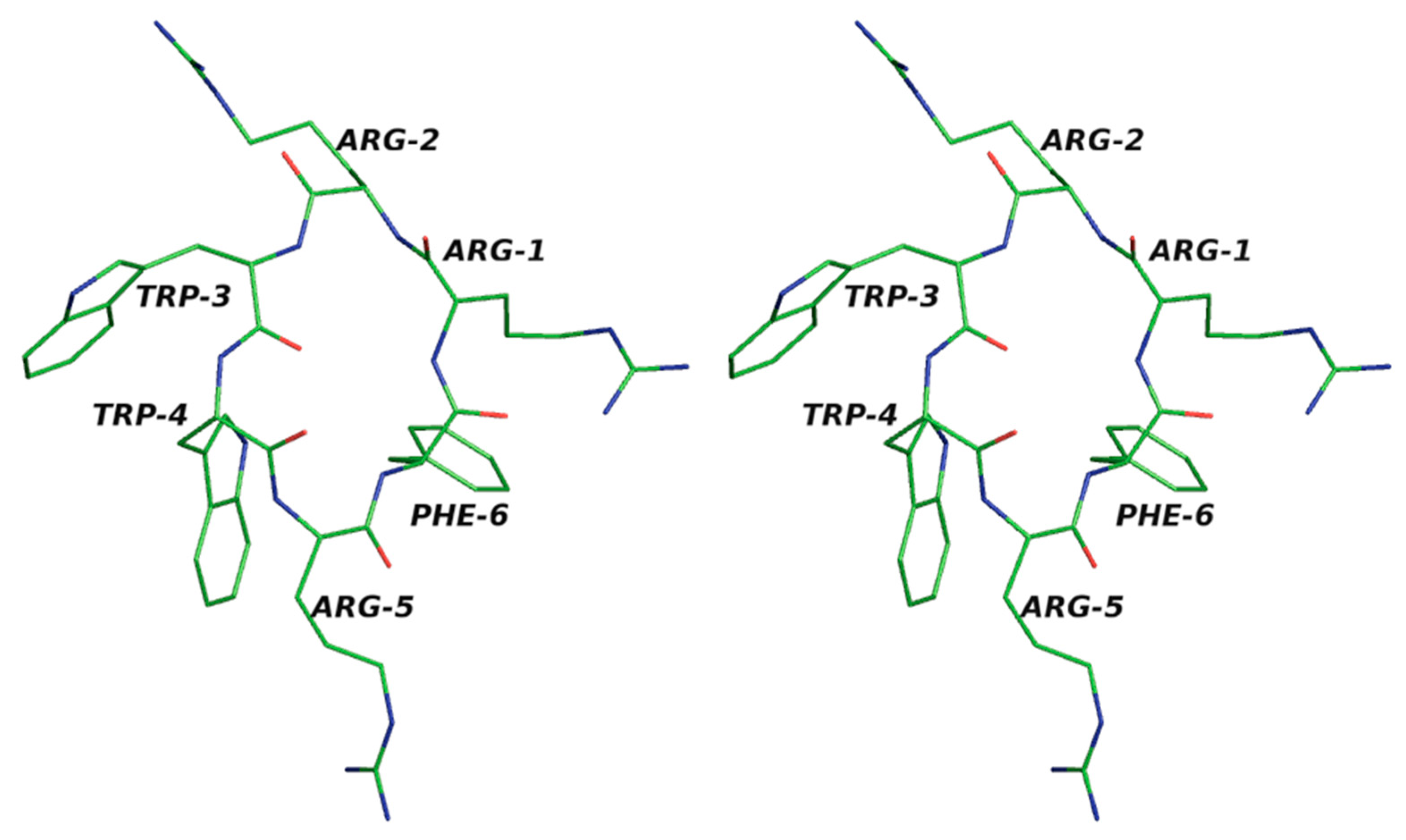
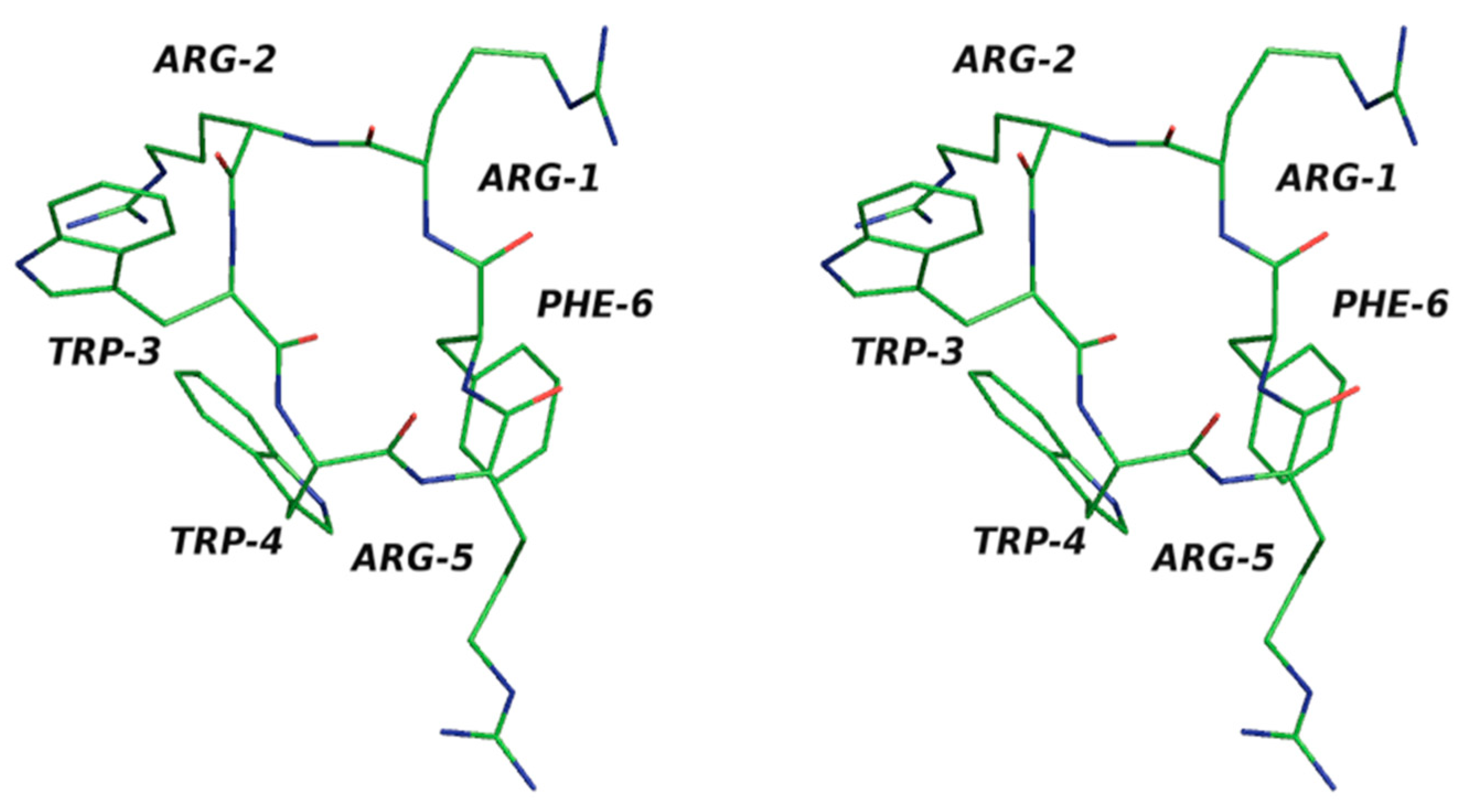
We investigated the cyclic hexapeptide of an antimicrobial peptide with the sequence RRWWRF in the current study. Here, the amino acid residues do not have similar patterns, and so we took this as a test case. We used the solved NMR structure from the PDB databank with id 1QVL [
13]. We took the best model as the input for our MOLS algorithm. To investigate conformational variations, we explored the conformation of this peptide in a vacuum and with an explicit solvent. This served as a preliminary study for our mean field technique, MOLS. We present a comprehensive account of the peptide’s conformational energy landscape, meticulously detailing its features within an explicit water environment. We also describe the various secondary motif structures and the backbone hydrogen bonding patterns, along with their effects on the environment. The Materials and Methods Section describes the addition of explicit water molecules and interaction energy calculations. Then, the results obtained for each are compared.
2. Materials and Methods
One of the methods used to thoroughly investigate the conformational space of oligopeptides is the mean field technique (MFT) when it is combined with MOLS sampling. The mean field technique (MFT) along with MOLS sampling was previously employed by researchers on the ECEPP3/and AMBER force fields, but the inclusion of solvent molecules was disregarded. A detailed explanation of this method was already outlined in [
14,
15] and is also available at
https://shodhganga.inflibnet.ac.in/handle/10603/198217, accessed on 18 April 2025. This method’s application in this study is described as follows.
This technique divides the conformational space into a number of subspaces, each containing numerous states. The effective potential obtained by setting a unique subspace to a particular state is conducted by taking the probability weighted average of the pairwise interactions in the subspace and comparing it to that of the states in all other subspaces. Initially, this method sets equal probabilities for all states within a given subspace; these probabilities are then recalculated using the Boltzmann function of the effective potential. The next cycle involves recalculating effective potentials using the new probabilities. We repeat this iterative procedure until the probabilities converge. The subspace with the highest probability after convergence will be the optimum one in the conformational space. For example, if we apply this method to determine the protein backbone conformation, then each of the backbone torsion angles will be considered the subspace, and the values that it can attain will be from 0 to 360°. Other consecutive states in steps of 10° can be accessed, but the effective potential cannot simply be calculated as the weighted sum of pairwise interactions because one of the torsion angles is set to such a value. If one sets the values for a pair of torsion angles, the occurring interactions will also depend on all the intermediate torsion angles. Calculating the potential is rigorous and requires considering all torsion angles, leading to a combinatorial explosion [
14,
15].
Using MOLS sampling, which tackles the small conformational space sample size, simplifies calculating the effective potential, easing the complexity of the problem. This method is used in the design of agricultural and clinical trials [
16]. Usually, this involves identifying a small representative sample of a multivariable experimental space. We perform these calculations on this subspace instead of the whole space. A Latin square of order m is the arrangement of m symbols in an m × m square matrix in such a way that each symbol occurs exactly once in every row and once in every column [
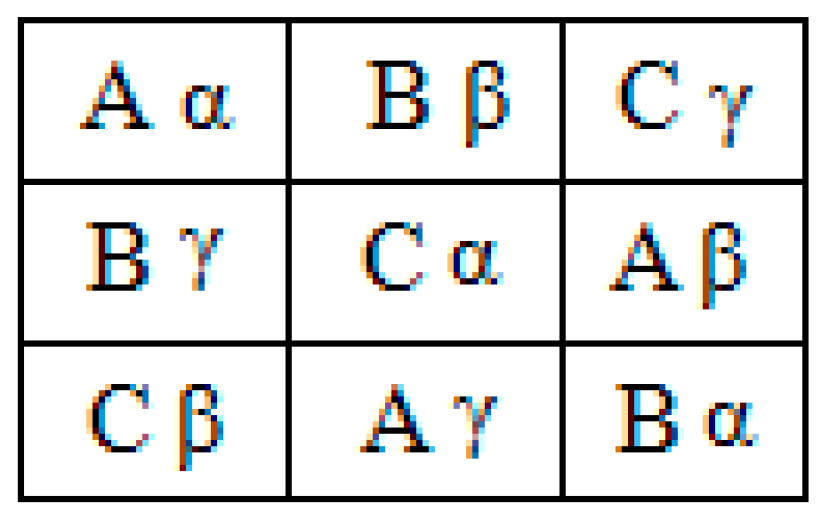
15]. A simple Latin square of order 3 is shown in
Figure 1. This example uses the letters of the Latin alphabet (A, B, C) as symbols to construct the square [
14,
15]. Two Latin squares of order m are called orthogonal if, when one is superimposed on the other, all m
2 possible combinations of the two sets of m symbols are present and each symbol of the one square occurs once and only once, with the symbols of the other squares being present.
Figure 2 shows a pair of orthogonal Latin squares involving two sets of three elements each [
14]. This pair was constructed by the superposition of two Latin squares, one square containing Latin letters (A, B, C) and the other constructed with Greek letters (α, β, γ).
Figure 2 shows that each subsquare contains one Latin and one Greek letter, and each letter combination appears only once. The symbols are arranged so that all m
2 pairs appear in the array [
14,
15]. Upon extending this concept to N mutually orthogonal Latin squares (MOLS), every pair is orthogonal. In the example of three MOLS (m = 3), the first square is orthogonal to the second and third squares, and the second is orthogonal to the third. The construction of a complete set of m-1 mutually orthogonal Latin squares of order n is shown to be possible if m is a prime number or an integer power of a prime [
14,
15,
16].
A single Latin square of order m allows one to investigate the effects of setting three subspaces, each at the m different states in only m
2 trials instead of the m
3 trials that would be required for a complete and exhaustive search of the sampling. A set of n mutually orthogonal Latin squares of order m allows one to investigate the effects of setting n factors to m different values, again in only m
2 (instead of m
n) trials. The above description shows that a distinctive feature of using MOLS to select sample points is the sample’s inclusion of every possible pairwise setting of all states of all subspaces [
14,
15,
16].
When applying this method to the exploration of conformational spaces, including the protein mapped out by the backbone torsion angles of a peptide chain, we identify subspaces with torsion angles, where there are n such angles, as well as identifying states as the values that the torsion angles can take, where there are m such values accessible to each torsion angle. Out of a total of m
n points available in the conformational space, we select n
2 points using MOLS and calculate the potential energy corresponding to each of these conformations. In order to calculate the effective potential realized from setting the torsion angle to a specific value, we take the Boltzmann-weighted average of the potential energies at these points in the MOLS grid at which the set value of the torsion angle appears. We note that, as described above, this ensures the pairwise sampling of the set value of the torsion angle with every value of every other torsion angle. We repeat this procedure for all values of all torsion angles, using the same MOLS grid specifying the m
2 conformations. At the end of this set of calculation, we obtain the effective potential corresponding to every set value of each torsion angle. In the routine MFT procedure, these potentials are used to recalculate probabilities, which are used to recalculate the effective potential in iterative cycles until it converges [
14]. In the present method using MOLS sampling, however, this is not possible since the effective potential is itself calculated as a Boltzmann-weighted average of the potential calculated in the MOLS grid. Thus, we use the effective potentials at the end of one round of calculations described above directly as substitutes for the final probabilities. For each torsion angle, we consider the value with the lowest effective potential the most probable one; the set of the most probable values for all torsion angles defines the optimum conformation of the peptides [
14,
15].
A crucial assumption underlying the MFT procedure, in particular when used with MOLS sampling, is regarding the degree of independence of the variables. The procedure works best when the variables are completely independent of each other; however, in that case, they are not required, and a few simple one-dimensional searches would quickly identify the optimum.
Backbone torsion angles are highly interdependent, and the procedure outlined above may not lead always to a single global optimum. The second assumption implicit in the procedure is that such a single global optimum exists on the potential energy landscape. In fact, as many studies have shown, most definitions of the force field operating in proteins and peptides describe the presence of several optima (minima) in the conformational space with approximately equal energy values. This method is an efficient way of exploring the entire space in an exhaustive manner and identifying all the optima. The construction of a single MOLS grid of order m, as described above, results in one optimal or low-energy conformation. Repeating this procedure by using a unique set of MOLS leads to another set. There are (m!)
n different ways of constructing the MOLS grid [
15]; in principle, the calculations may be repeated as many times as possible to repeatedly obtain all low-energy conformations. This does not mean that there are (m!)
n low-energy conformations for a peptide. The conformations generated in the preceding cycles are obtained as they were in the previous process.
The well-known Met-enkephalin test case proved the usefulness of this method; the procedure, repeated over a few hundred cycles, exhaustively identified all minima. The energies of the experimental structure and the theoretical structure also fall within the range of the energies generated by this method. Some structures were also present with an energy that had not been seen anywhere else before. In the abovementioned method, we used the ECEPP/3 semi-empirical force field, without including explicit water molecules [
14,
15].
In the present work, we implemented our method for cyclic peptides. Here, we used the input as the selected structure. The program was changed so that the user could provide their own built structure as the input. Here, we used the antimicrobial cyclic hexapeptide PDB ID 1QVL as the test case [
13]. The cyclic nature of the peptide backbones means that the first and last terminals of the linear peptide fuse to form a peptide bond, eliminating main chain rotation. We applied rotations to the side chains and then generated an optimized structure using two different environments, in the presence and in the absence of an explicit solvent. We used the VMD program [
17] to explore both environments. We added explicit water molecules to each of the defined n2 MOLS conformations by placing a solvent box using the VMD program. We solvated the peptide using TIP3P water molecules, extending the water approximately 10 Å in each direction from the molecule’s ends. When the side chains of the peptide were in or out of plane, it was ensured that the solvent molecule always covered them. After this, we calculated the conformational energy of the peptide structure using the CHARMM22 potential [
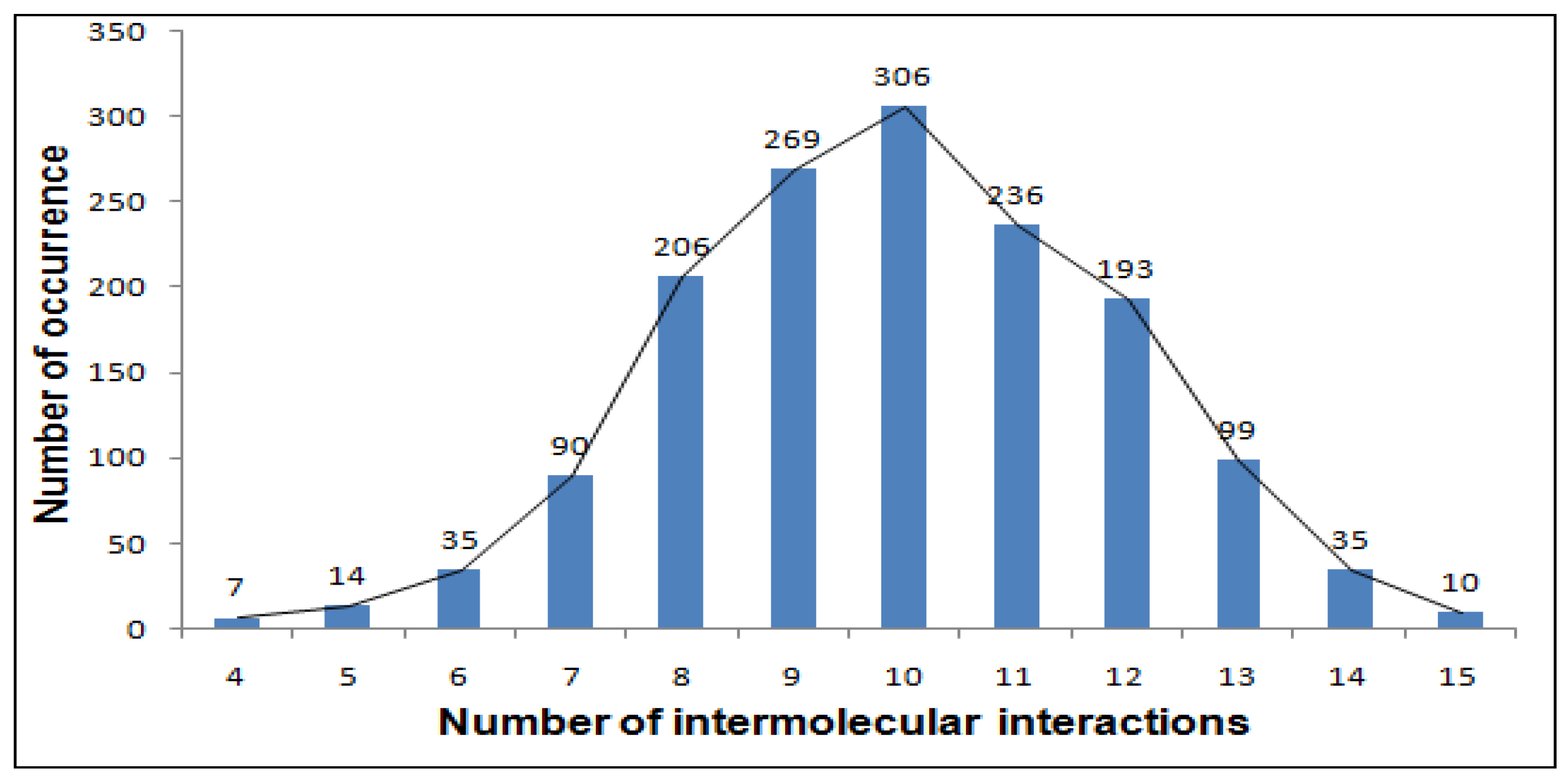
18]. The calculation of the total energy involved the summation of several energy terms, namely, the torsion angle energy, the energy from electrostatic interactions, the van der Waals energy, and the energy associated with intermolecular interactions. The MOLS method involved obtaining effective energies for each conformational variable value based on the calculated potential energy of the n
2 molecule, in order to identify the optimal conformation associated with it. We solvated the obtained optimal conformation using the same procedure and removed the conformational constraints by performing 1000 steps of conjugate gradient minimization using the NAMD minimization procedure [
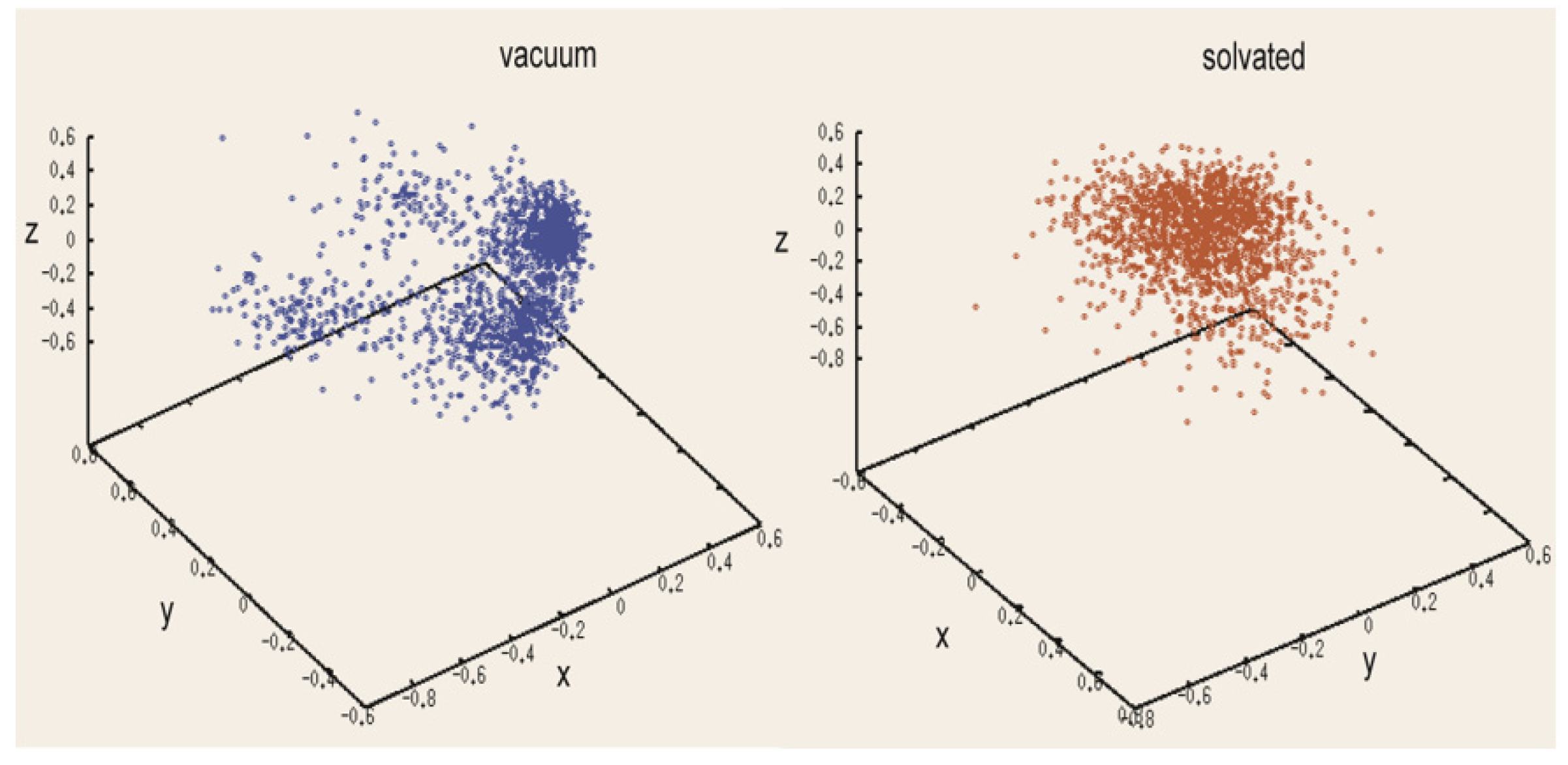
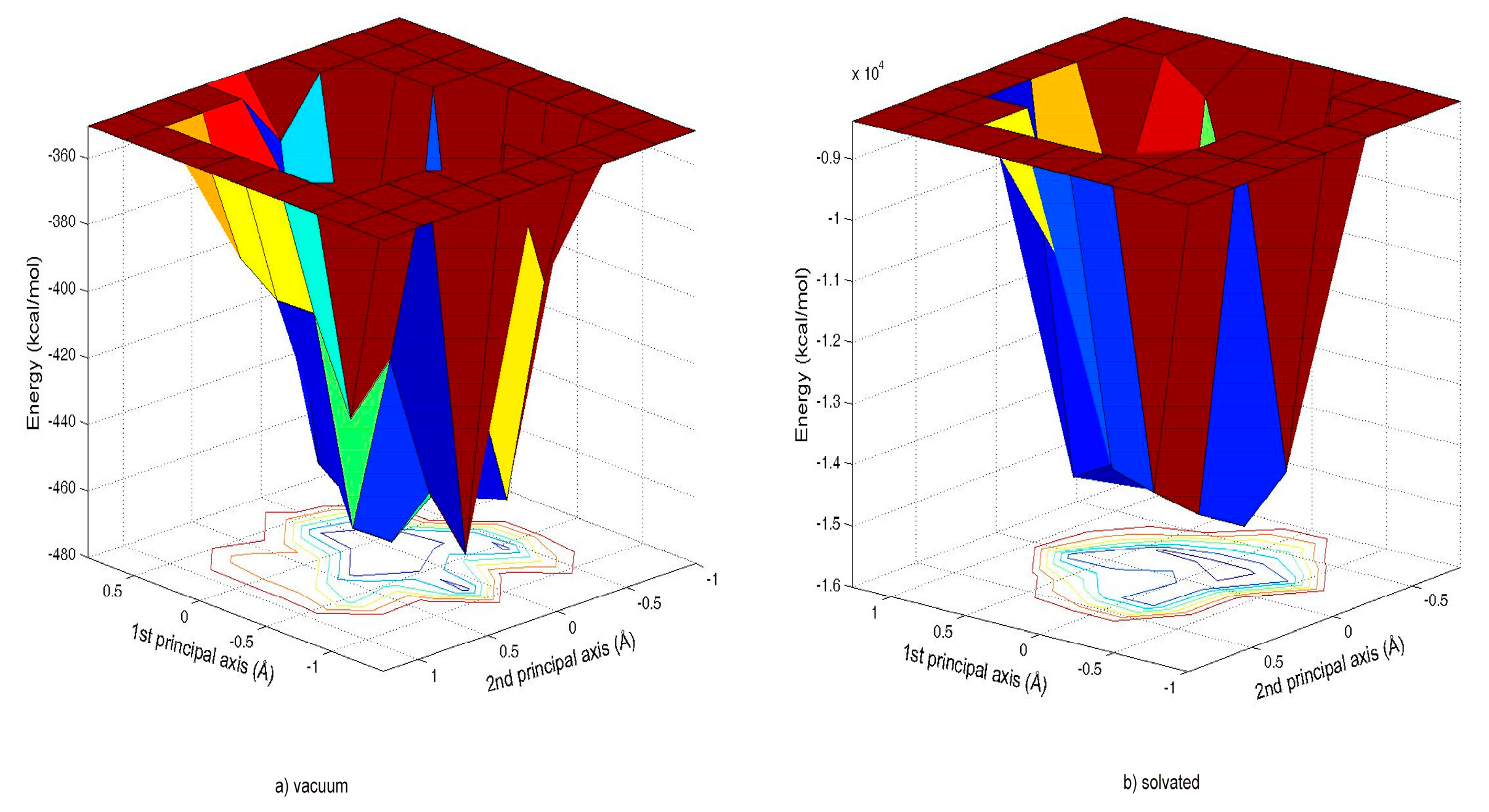
19]. In the minimization process, the energies of bond length, bond angle, and torsion angle and the improper nonbonded energies were taken into account. We did not use water–water interaction terms. Furthermore, a separate set of 1500 conformations was generated for the peptide, which was devoid of water molecules and subjected to a vacuum environment. Computation was conducted to generate one structure with and without a solvent using an AMD Opteron processor at 2.2 GHz for 57 min and 10 min, respectively.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}