
Theoretical Study of CO Oxidation on Pt Single-Atom Catalyst Decorated C3N Monolayers with Nitrogen Vacancies

 , , and
, , and
Abstract

1. Introduction
2. Computational Method
3. Results
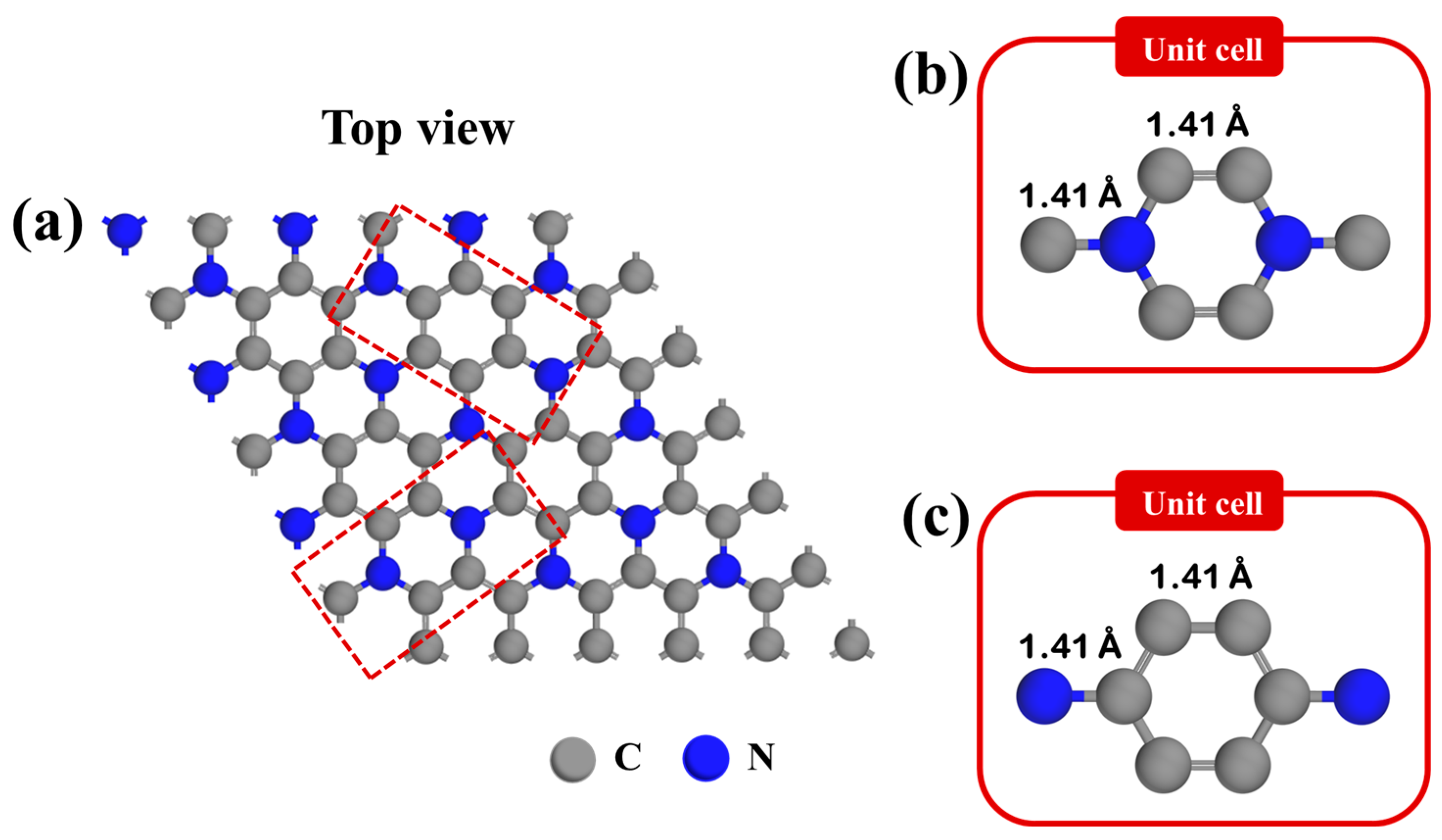
3.1. Structural Model of Pristine C3N
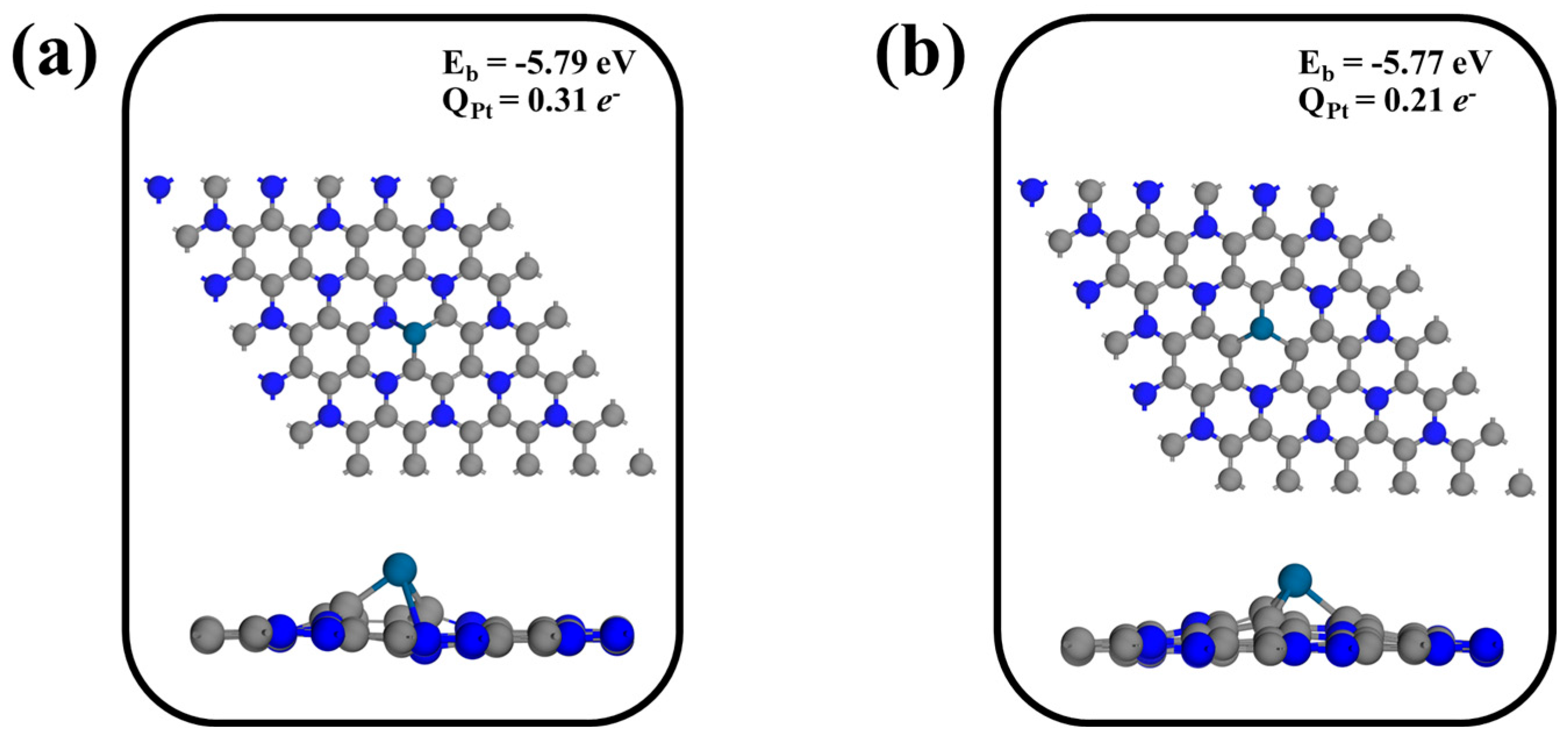
3.2. The Defective Structure of C3N
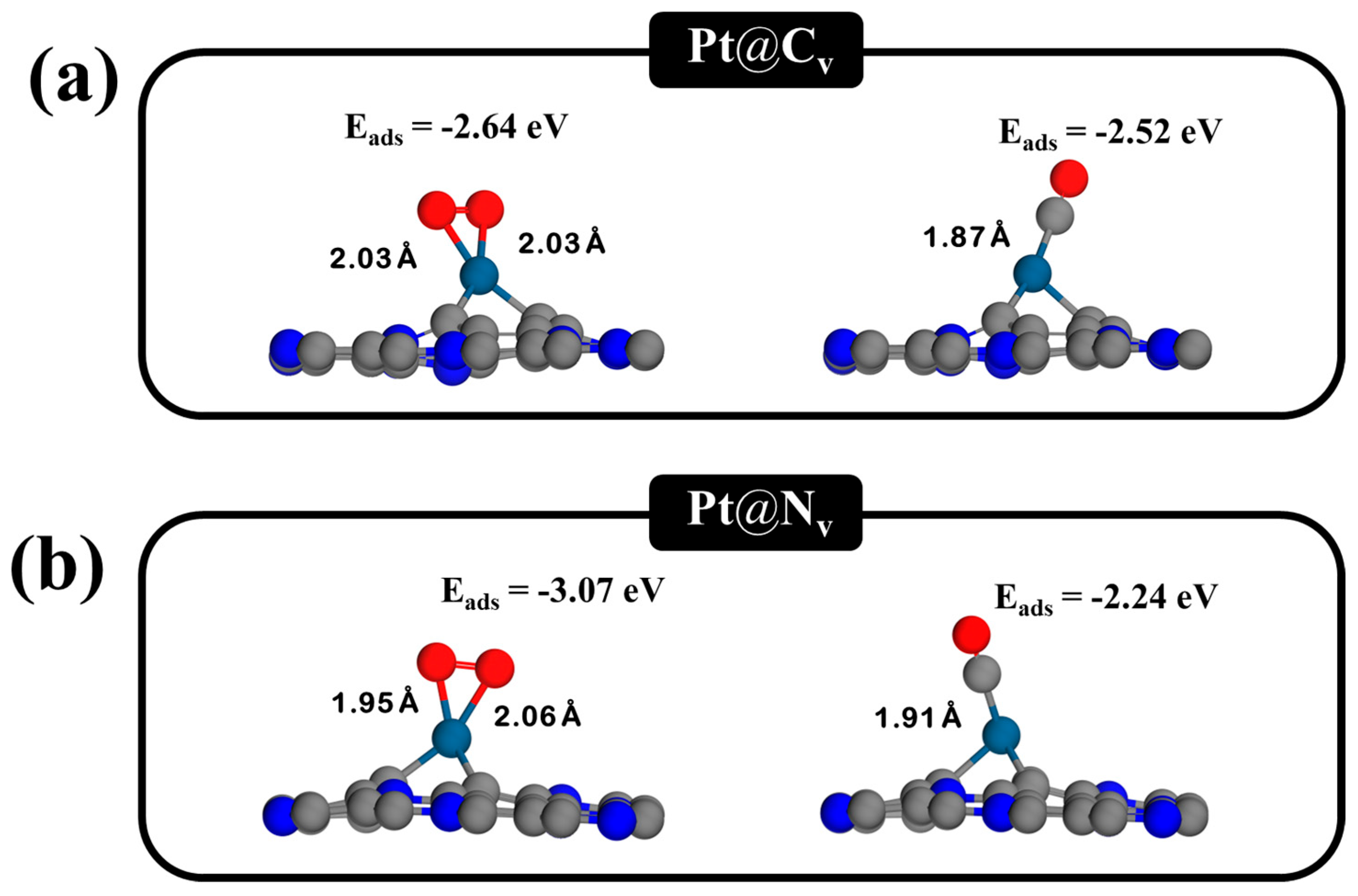
3.3. The O2 and CO Adsorption on Pt-Doped C3N Monolayers with Single Vacancies
3.4. Reaction Mechanism of CO Oxidation on Pt@NV-C3N
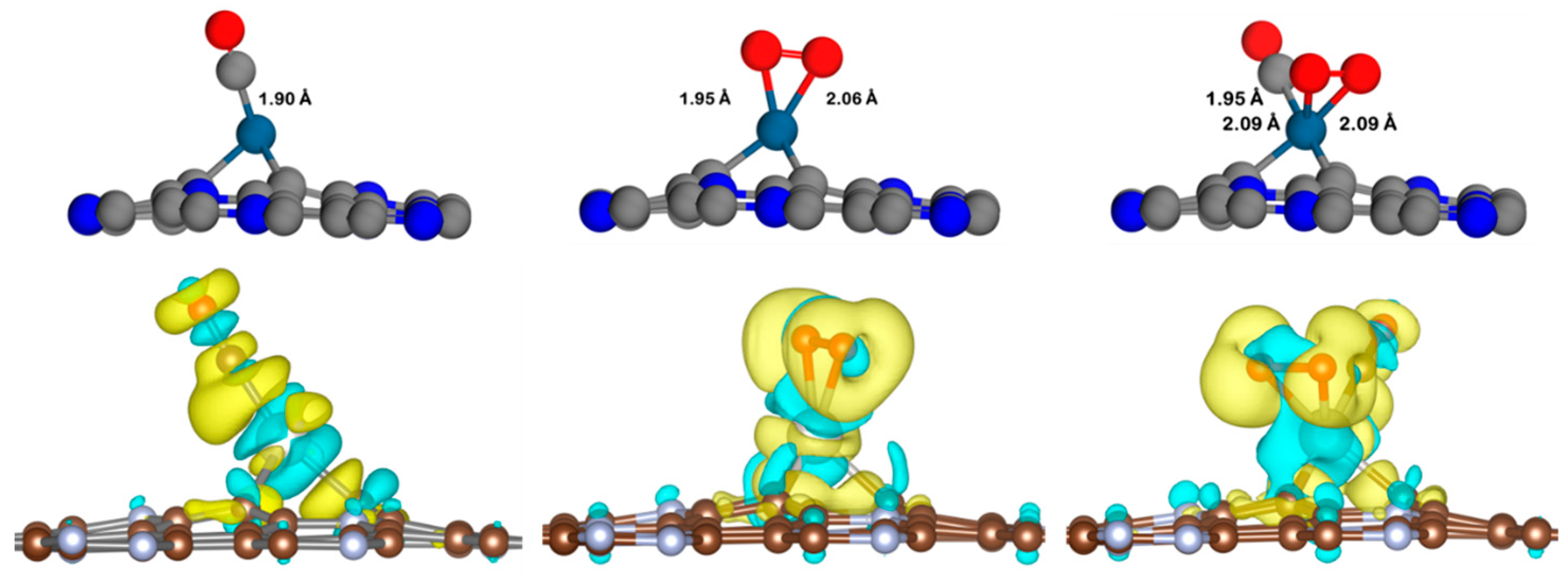
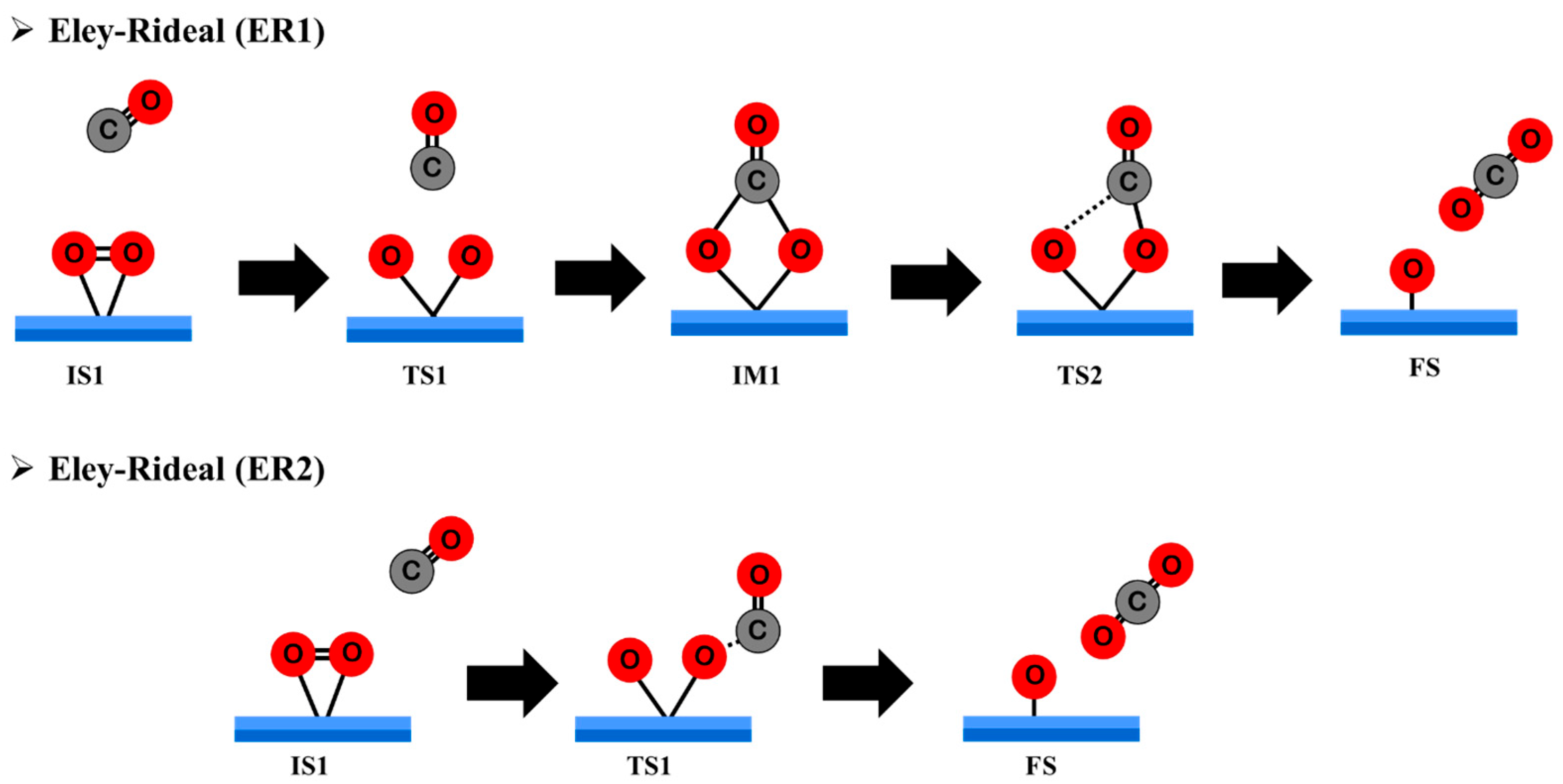
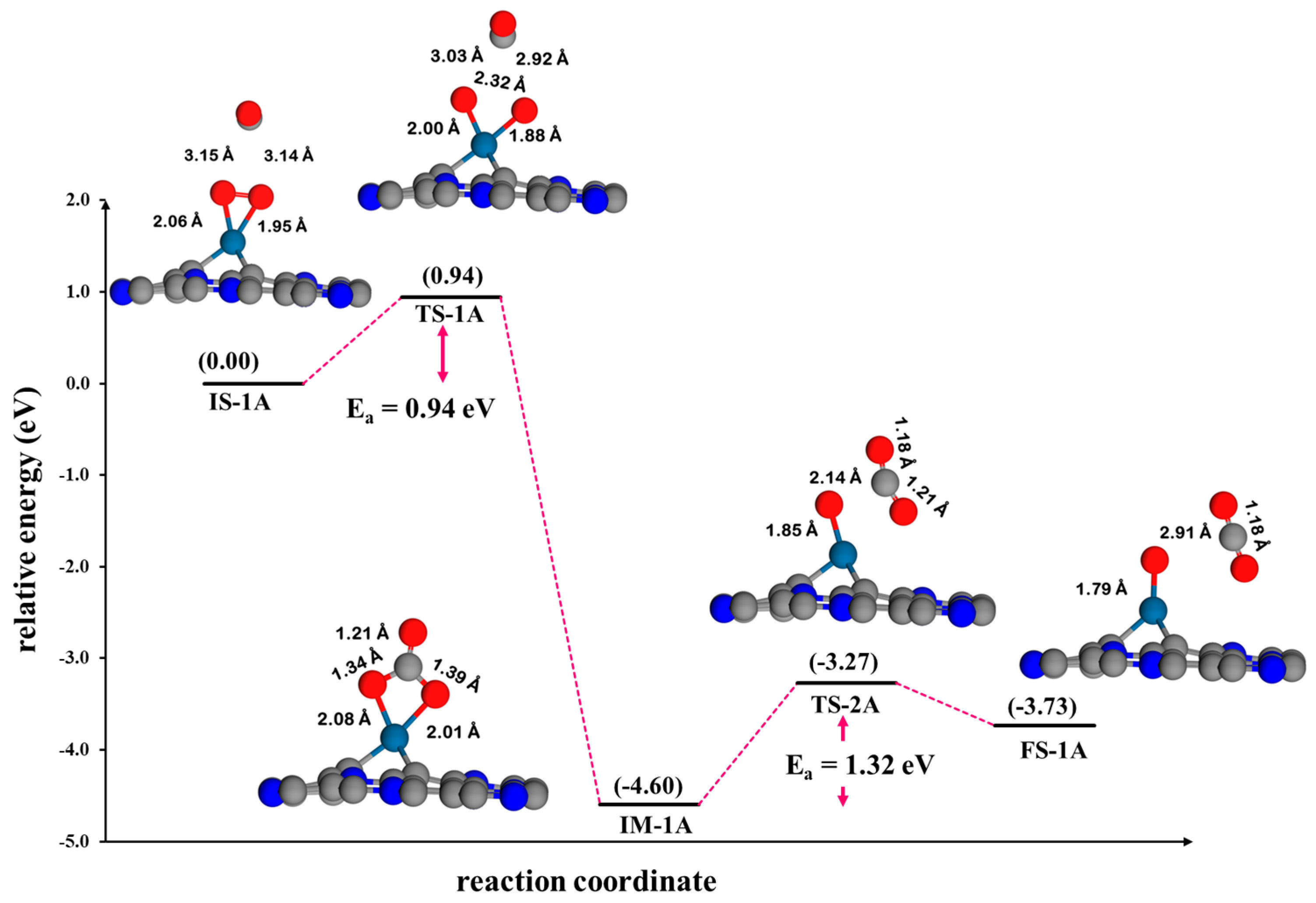
3.4.1. Eley–Rideal Mechanism (ER) on Pt@NV-C3N
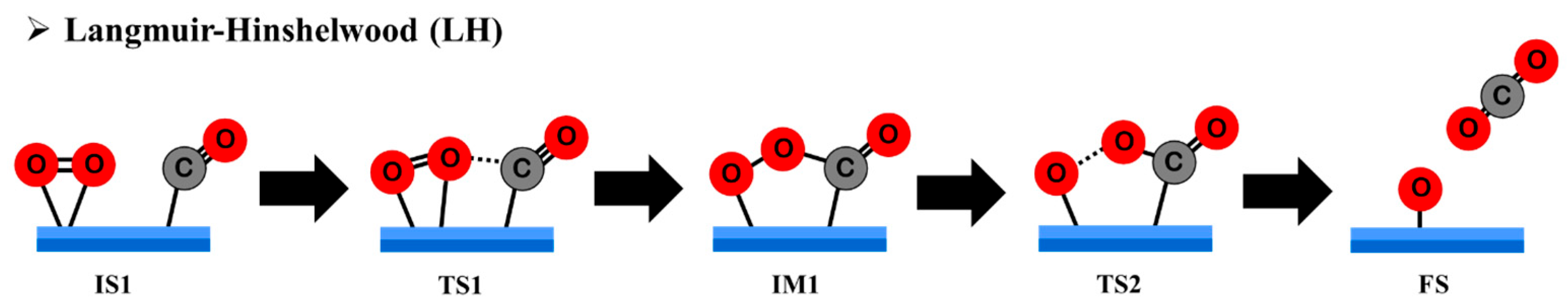
3.4.2. Langmuir–Hinshelwood Mechanism (LH) on Pt@NV-C3N
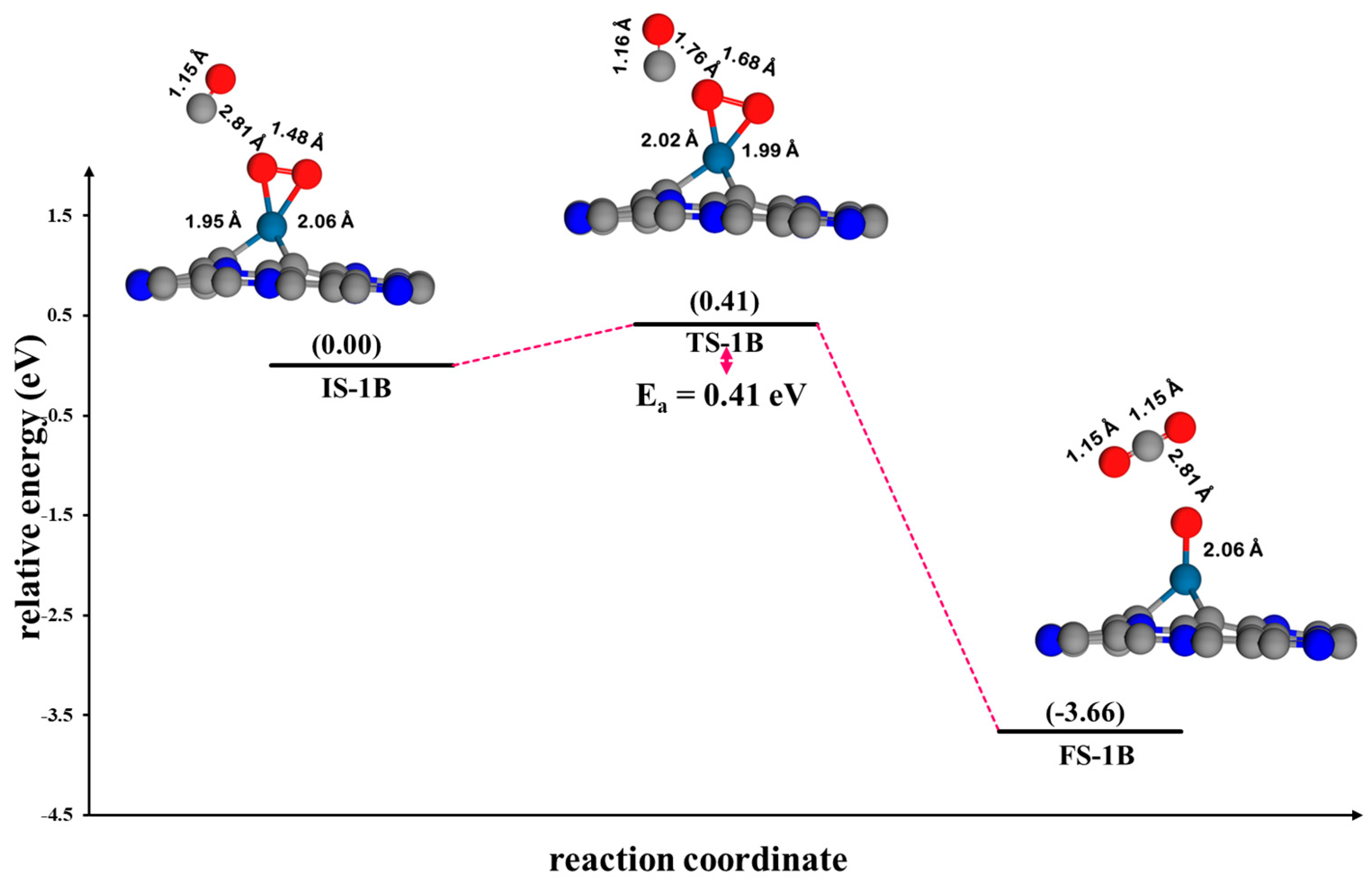
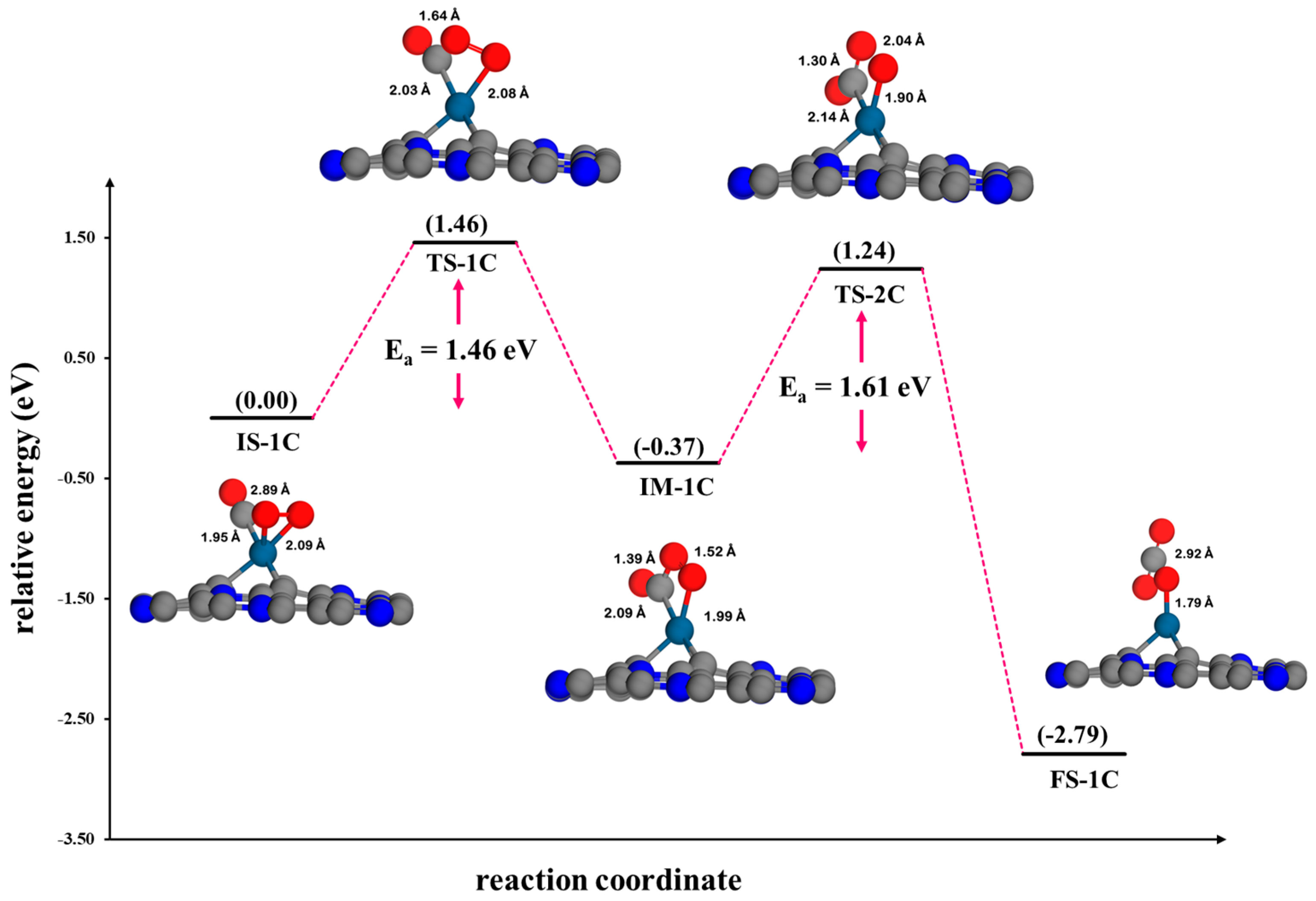
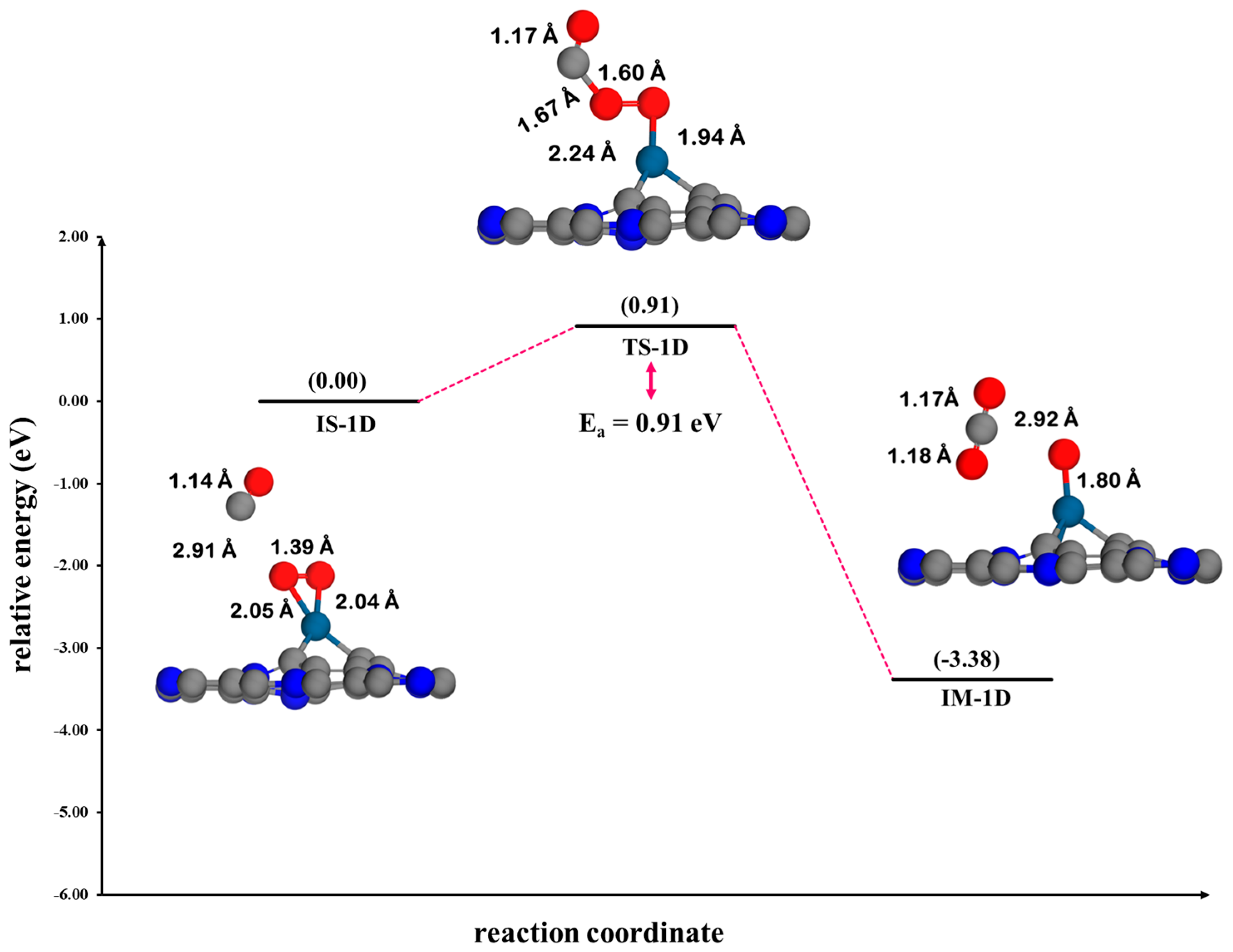
3.4.3. Eley–Rideal Mechanism (ER2) on Pt@CV-C3N
4. Discussion
4.1. Comparison of the Reaction Mechanism of CO Oxidation on Pt@NV-C3N
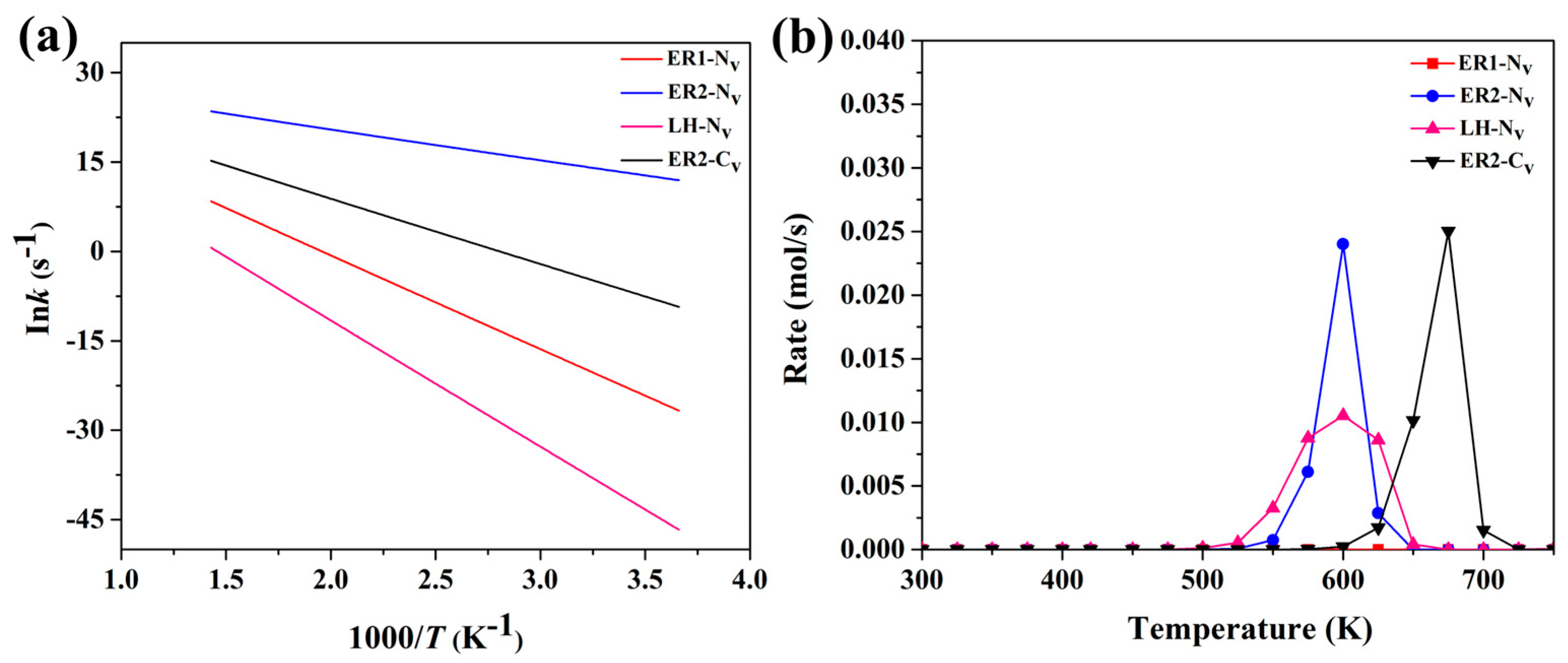
4.2. Kinetic Studies and Microkinetic Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density functional theory |
| LH | Langmuir–Hinshelwood |
| ER | Eley–Rideal |
| Ea | Activation energy barriers |
References
- Esrafili, M.D.; Heydari, S. B-doped C3N monolayer: A robust catalyst for oxidation of carbon monoxide. Theor. Chem. Acc. 2019, 138, 57. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P. Boosting graphene reactivity with co-doping of boron and nitrogen atoms: CO oxidation by O2 molecule. Appl. Surf. Sci. 2018, 455, 808–814. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P. A DFT study on the possibility of using a single Cu atom incorporated nitrogen-doped graphene as a promising and highly active catalyst for oxidation of CO. Int. J. Quantum Chem. 2019, 119, e25857. [Google Scholar] [CrossRef]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO oxidation as a prototypical reaction for heterogeneous processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, J.; Chen, Z. Fe-anchored graphene oxide: A low-cost and easily accessible catalyst for low-temperature CO oxidation. J. Phys. Chem. C 2012, 116, 2507–2514. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X. The theoretical study of Rh single atom catalysts decorated C3N monolayer with N vacancy for CO oxidations. Appl. Phys. A 2023, 129, 325. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic oxidation of carbon monoxide over transition metal oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y. A comparative study of CO oxidation on Cu-doped C3N monolayer with N and C vacancies. J. Phys. Chem. Solids 2023, 180, 111441. [Google Scholar] [CrossRef]
- Prasad, R.; Singh, P. A review on CO oxidation over copper chromite catalyst. Catal. Rev. 2012, 54, 224–279. [Google Scholar] [CrossRef]
- Kim, H.J.; Jang, M.G.; Shin, D.; Han, J.W. Design of ceria catalysts for low-temperature CO oxidation. ChemCatChem 2020, 12, 11–26. [Google Scholar] [CrossRef]
- Wallander, H.J.; Gajdek, D.; Albertin, S.; Harlow, G.; Braud, N.; Buß, L.; Krisponeit, J.-O.; Flege, J.I.; Falta, J.; Lundgren, E.; et al. Dynamic Behavior of Tin at Platinum Surfaces during Catalytic CO Oxidation. ACS Catal. 2023, 13, 16158–16167. [Google Scholar] [CrossRef]
- Zagaynov, I.V.; Liberman, E.Y.; Naumkin, A.V. Influence of Pt/Pd state on ceria-based support in CO oxidation. J. Rare Earths 2023, 41, 1963–1968. [Google Scholar] [CrossRef]
- Xu, L.; Pan, Y.; Li, H.; Xu, R.; Sun, Z. Highly active and water-resistant Lanthanum-doped platinum-cobalt oxide catalysts for CO oxidation. Appl. Catal. B Environ. 2023, 331, 122678. [Google Scholar] [CrossRef]
- Chen, C.; Chen, J.-L.; Feng, L.; Hu, J.; Chai, X.; Liu, J.-X.; Li, W.-X. Reactant-Induced Dynamic Stabilization of Highly Dispersed Pt Catalysts on Ceria Dictating the Reactivity of CO Oxidation. ACS Catal. 2024, 14, 3504–3513. [Google Scholar] [CrossRef]
- Feng, C.; Liu, X.; Zhu, T.; Tian, M. Catalytic oxidation of CO on noble metal-based catalysts. Environ. Sci. Pollut. Res. 2021, 28, 24847–24871. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.; Lai, C.; Zhang, Y.; Lin, X.; Ding, S. Recent advances in noble metal-based catalysts for CO oxidation. RSC Adv. 2024, 14, 30566–30581. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Gomes, J.R.B. DFT Study of the CO Oxidation on the Au (321) Surface. J. Phys. Chem. C 2008, 112, 17291–17302. [Google Scholar] [CrossRef]
- Rao, X.; Si, Q.; Shi, T.; Han, X.; Ma, S. Fe-doped C3N monolayer as a promising SAC for CO oxidation with low temperature and high reactivity. Comput. Theor. Chem. 2021, 1194, 113080. [Google Scholar] [CrossRef]
- Bafekry, A.; Ghergherehchi, M.; Shayesteh, S.F.; Peeters, F. Adsorption of molecules on C3N nanosheet: A first-principles calculations. Chem. Phys. 2019, 526, 110442. [Google Scholar] [CrossRef]
- Cui, H.; Liu, Z.; Jia, P. Pd-doped C3N monolayer: A promising low-temperature and high-activity single-atom catalyst for CO oxidation. Appl. Surf. Sci. 2021, 537, 147881. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Heydari, S. An effective approach for tuning catalytic activity of C3N nanosheets: Chemical-doping with the Si atom. J. Mol. Graph. Model. 2019, 92, 320–328. [Google Scholar] [CrossRef]
- Meng, Y.; Gao, Y.; Li, K.; Tang, H.; Wang, Y.; Wu, Z. Transition metal doped C3N monolayer as efficient electrocatalyst for carbon dioxide electroreduction: A computational study. Appl. Surf. Sci. 2021, 542, 148568. [Google Scholar] [CrossRef]
- Li, X.; Guo, T.; Zhu, L.; Ling, C.; Xue, Q.; Xing, W. Charge-modulated CO2 capture of C3N nanosheet: Insights from DFT calculations. Chem. Eng. J. 2018, 338, 92–98. [Google Scholar] [CrossRef]
- Xu, G.; Wang, R.; Yang, F.; Ma, D.; Yang, Z.; Lu, Z. CO oxidation on single Pd atom embedded defect-graphene via a new termolecular Eley-Rideal mechanism. Carbon 2017, 118, 35–42. [Google Scholar] [CrossRef]
- Xu, G.; Wang, R.; Ding, Y.; Lu, Z.; Ma, D.; Yang, Z. First-principles study on the single Ir atom embedded graphdiyne: An efficient catalyst for CO oxidation. J. Phys. Chem. C 2018, 122, 23481–23492. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, Z.; Xu, G.; Wang, T.; Ma, D.; Yang, Z.; Yang, L. Single Pt atom stabilized on nitrogen doped graphene: CO oxidation readily occurs via the tri-molecular Eley—Rideal mechanism. Phys. Chem. Chem. Phys. 2015, 17, 20006–20013. [Google Scholar] [CrossRef]
- González-Torres, J.C.; Bertin, V.; Poulain, E.; Olvera-Neria, O. The CO oxidation mechanism on small Pd clusters. A theoretical study. J. Mol. Model. 2015, 21, 279. [Google Scholar] [CrossRef]
- Baskaran, S.; Jung, J. Termolecular Eley–Rideal pathway for efficient CO oxidation on phosphorene-supported single-atom cobalt catalyst. Bull. Korean Chem. Soc. 2022, 43, 1254–1261. [Google Scholar] [CrossRef]
- Luo, M.; Liang, Z.; Chen, M.; Liu, C.; Qi, X.; Peera, S.G.; Liu, J.; Liang, T. Theoretical investigation on catalytic mechanisms of oxygen reduction and carbon monoxide oxidation on the MnN x system. New J. Chem. 2020, 44, 15724–15732. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Li, Q.; Bai, X.; Wang, J. Metal single-atom coordinated graphitic carbon nitride as an efficient catalyst for CO oxidation. Nanoscale 2020, 12, 364–371. [Google Scholar] [CrossRef]
- Hussain, S.; Talib, S.H.; Shahzad, S.R.; Muhammad, S.; Mohamed, S.; Qurashi, A.; Wang, H.; Lu, Z. Heterogeneous Co1/P1Mo12O40 single-atom catalyst for CO oxidation via termolecular Eley-Rideal (TER) mechanism. Mol. Catal. 2023, 550, 113539. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J.; Hafner, J. Ab initio force constant approach to phonon dispersion relations of diamond and graphite. Europhys. Lett. 1995, 32, 729. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Först, C.J.; Schimpl, J. Projector augmented wave method: Ab initio molecular dynamics with full wave functions. Bull. Mater. Sci. 2003, 26, 33–41. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 54104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Z.; Qin, Z.; Zhang, G.; Yue, H.; Liang, B.; Luo, D. Dissolution behavior and mechanism of low valence vanadium of vanadium-iron spinel in sulfuric acid solution. J. Mater. Res. Technol. 2021, 12, 1391–1402. [Google Scholar] [CrossRef]
- Kilinc, D. Co complex modified on Eupergit C as a highly active catalyst for enhanced hydrogen production. Int. J. Hydrogen Energy 2022, 47, 11894–11903. [Google Scholar] [CrossRef]
- Lv, J.; Hong, J.; Liang, B.; Zhao, E.; Zeng, K.; Chen, M.; Hu, J.; Yang, G. Study of the curing kinetics of melamine/phthalonitrile resin system. Thermochim. Acta 2020, 683, 178442. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Xue, S.; Wu, Y.; Li, Z.; Chang, L. Evaluation of the susceptibility of coal to spontaneous combustion by a TG profile subtraction method. Korean J. Chem. Eng. 2016, 33, 862–872. [Google Scholar] [CrossRef]
- Zhang, X.; Bi, F.; Zhu, Z.; Yang, Y.; Zhao, S.; Chen, J.; Lv, X.; Wang, Y.; Xu, J.; Liu, N. The promoting effect of H2O on rod-like MnCeOx derived from MOFs for toluene oxidation: A combined experimental and theoretical investigation. Appl. Catal. B Environ. 2021, 297, 120393. [Google Scholar] [CrossRef]
- Jiang, Q.; Huang, M.; Qian, Y.; Miao, Y.; Ao, Z. Excellent sulfur and water resistance for CO oxidation on Pt single-atom-catalyst supported by defective graphene: The effect of vacancy type. Appl. Surf. Sci. 2021, 566, 150624. [Google Scholar] [CrossRef]
- Wu, Q.; Hu, Q.; Hou, Y.; Wang, H.; Zhou, A.; Wang, L.; Cao, G. Unexpected ground-state structures and properties of carbon nitride C3N at ambient and high pressures. Mater. Des. 2018, 140, 45–53. [Google Scholar] [CrossRef]
- Wang, H.; Wu, H.; Yang, J. C3N: A two dimensional semiconductor material with high stiffness, superior stability and bending Poisson’s effect. arXiv 2017, arXiv:1703.08754. [Google Scholar]
- Sakhraoui, T. Effect of vacancy defect and strain on the structural, electronic and magnetic properties of carbon nitride 2D monolayers by DFTB method. J. Phys. Condens. Matter 2023, 35, 324003. [Google Scholar] [CrossRef]
- Makaremi, M.; Mortazavi, B.; Singh, C.V. Adsorption of metallic, metalloidic, and nonmetallic adatoms on two-dimensional C3N. J. Phys. Chem. C 2017, 121, 18575–18583. [Google Scholar] [CrossRef]
- Mizuno, S.; Fujita, M.; Nakao, K. Electronic states of graphitic heterocompounds of carbon, boron and nitrogen. Synth. Met. 1995, 71, 1869–1870. [Google Scholar] [CrossRef]
- Mortazavi, B. Ultra high stiffness and thermal conductivity of graphene like C3N. Carbon 2017, 118, 25–34. [Google Scholar] [CrossRef]
- Ragab, A.H.; Khan, I.; Khan, M.; Yousef, T.A.; Abou-Krisha, M.M.; Kenawy, S.H.; Ansari, M.Z. Efficient oxidation of CO over highly active Al-decorated nitrogenated holey 2D-graphene: A DFT perspective. Inorg. Chem. Commun. 2023, 155, 110977. [Google Scholar] [CrossRef]
- Chang, F.; Li, J.; Kou, Y.; Bao, W.; Shi, Z.; Zhu, G.; Kong, Y. The intense charge migration and efficient photocatalytic NO removal of the S-scheme heterojunction composites Bi7O9I3-BiOBr. Sep. Purif. Technol. 2025, 353, 128402. [Google Scholar] [CrossRef]
- Luo, M.; Liang, Z.; Chen, M.; Peera, S.G.; Liu, C.; Yang, H.; Qi, X.; Liu, J.; Liang, T. Catalytic oxidation mechanisms of carbon monoxide over single-and double-vacancy Mn-embedded graphene. New J. Chem. 2020, 44, 9402–9410. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, J.; Ao, Z.; Huang, H.; He, H.; Wu, Y. First principles study on the CO oxidation on Mn-embedded divacancy graphene. Front. Chem. 2018, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yu, Q.; Yang, X.; Zhang, T.; Li, J. A systematic theoretical study on FeOx-supported single-atom catalysts: M1/FeOx for CO oxidation. Nano Res. 2018, 11, 1599–1611. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Chen, Z. 1 + 1′ > 2: Heteronuclear biatom catalyst outperforms its homonuclear counterparts for CO oxidation. Small Methods 2019, 3, 1800480. [Google Scholar] [CrossRef]
- Luo, M.; Liang, Z.; Liu, C.; Qi, X.; Chen, M.; Sagar, R.U.R.; Yang, H.; Liang, T. Single–atom manganese and nitrogen co-doped graphene as low-cost catalysts for the efficient CO oxidation at room temperature. Appl. Surf. Sci. 2021, 536, 147809. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, Q.; Liu, J.; Lu, Z. The CO oxidation on Mn atom embedded in N vacancy of C3N monolayer: A DFT-D study. Surf. Sci. 2023, 734, 122320. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Fang, S.; Yao, S. Revealing the differences in CO oxidation activity of Fe-CeO2, Fe2O3, and CeO2 using operando CO2-DRIFTS-MS: Carbonate species and desorption process. J. Alloys Compd. 2025, 1010, 177414. [Google Scholar] [CrossRef]
- Al Soubaihi, R.M.; Saoud, K.M.; Myint, M.T.Z.; Göthelid, M.A.; Dutta, J. CO oxidation efficiency and hysteresis behavior over mesoporous Pd/SiO2 catalyst. Catalysts 2021, 11, 131. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Position | Type of CO Oxidation Reaction | Calculation |
|---|---|---|---|
| Si-C3N [21] | C vacancy | LH ( = 0.38 eV) | Dmol3 |
| B-C3N [1] | C vacancy | LH ( = 0.32 eV) | Dmol3 |
| Gr-BCN2 [2] | C vacancy | LH ( = 0.46 eV) | Dmol3 |
| Gr-BN3 [2] | C vacancy | LH ( = 0.01 eV) | Dmol3 |
| CuN3-Gr [3] | C vacancy | ER ( = 3.20 eV) | Dmol3 |
| Mn-Gr [59] | C vacancy | ER ( = 0.83 eV) | Dmol3 |
| Pd-C3N [20] | N vacancy | ER ( = 0.64 eV) | Dmol3 |
| Mn-C3N [60] | N vacancy | ER ( = 0.57 eV) | Dmol3 |
| Fe-C3N [18] | N vacancy | ER ( = 0.47 eV) | Dmol3 |
| Pt-C3N* | C vacancy | ER ( = 0.91 eV) | VASP |
| Pt-C3N* | N vacancy | ER ( = 0.41 eV) | VASP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamchompoo, S.; Injongkol, Y.; Yodsin, N.; Zhang, R.-Q.; Kunaseth, M.; Jungsuttiwong, S. Theoretical Study of CO Oxidation on Pt Single-Atom Catalyst Decorated C3N Monolayers with Nitrogen Vacancies. Sci 2025, 7, 101. https://doi.org/10.3390/sci7030101
Kamchompoo S, Injongkol Y, Yodsin N, Zhang R-Q, Kunaseth M, Jungsuttiwong S. Theoretical Study of CO Oxidation on Pt Single-Atom Catalyst Decorated C3N Monolayers with Nitrogen Vacancies. Sci. 2025; 7(3):101. https://doi.org/10.3390/sci7030101
Chicago/Turabian StyleKamchompoo, Suparada, Yuwanda Injongkol, Nuttapon Yodsin, Rui-Qin Zhang, Manaschai Kunaseth, and Siriporn Jungsuttiwong. 2025. "Theoretical Study of CO Oxidation on Pt Single-Atom Catalyst Decorated C3N Monolayers with Nitrogen Vacancies" Sci 7, no. 3: 101. https://doi.org/10.3390/sci7030101
APA StyleKamchompoo, S., Injongkol, Y., Yodsin, N., Zhang, R.-Q., Kunaseth, M., & Jungsuttiwong, S. (2025). Theoretical Study of CO Oxidation on Pt Single-Atom Catalyst Decorated C3N Monolayers with Nitrogen Vacancies. Sci, 7(3), 101. https://doi.org/10.3390/sci7030101