1. Introduction
Cholesterol is an essential lipid that plays a pivotal role in preserving cellular architecture and ensuring proper function in animal eukaryotic cells. As a key component of cell membranes, it regulates fluidity and stability, ensuring proper membrane organization and signaling [
1]. In the nervous system, it is vital for synapse formation, myelin sheath integrity [
2], and neurotransmission [
3]. However, cholesterol imbalance has been linked to a range of cardiovascular disorders and neurodegenerative diseases, including Alzheimer’s, Huntington’s, and Parkinson’s disease [
4]. Although extensive research has shed light on cholesterol’s intracellular distribution and trafficking, the exact mechanisms that govern these processes remain only partially understood [
5]. By elucidating these processes, we can uncover novel targets for therapeutic intervention, ultimately leading to more effective treatments. Additionally, a deeper understanding of cholesterol trafficking could reveal fundamental insights into cellular homeostasis and the pathology of diseases associated with lipid dysregulation, thereby advancing both basic science and clinical applications.
Fluorescent cholesterol probes [
6] and sensors [
7,
8] are indispensable tools for studying cholesterol trafficking in live cells because they enable real-time visualization, high-resolution tracking, and quantitative analysis of cholesterol dynamics. Unlike traditional biochemical methods, such as lipid extraction and mass spectrometry [
9], which require cell disruption and provide only static snapshots, fluorescent probes allow continuous monitoring of cholesterol movement in living systems.
Genetically encoded cholesterol sensors and chemical fluorescent probes each have unique advantages and limitations when studying cholesterol trafficking in live cells. Due to the unique nature of cholesterol, there is no universal fluorescent cholesterol probe. The choice between genetically encoded cholesterol reporters and chemical fluorescent probes depends on the research question [
10].
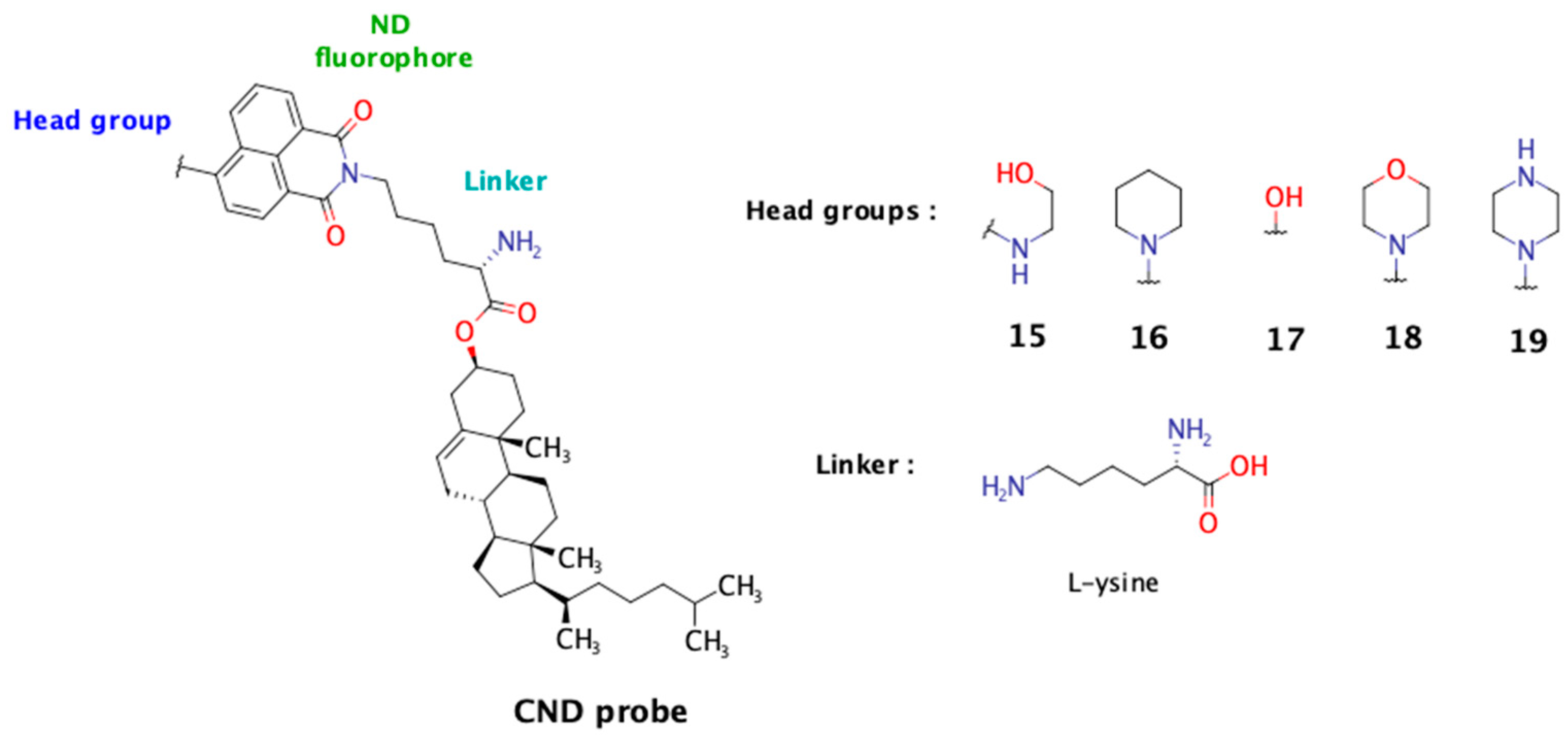
Among existing fluorescent cholesterol probes, the fluorescent cholesteryl probes based on 1,8-naphthalimide scaffold (CNDs) recently developed in our group demonstrate excellent solvatochromic properties, partitioning into Lo phases in model membranes, and some of them are also pH sensitive [
11]. The modular structure of these probes allows us to investigate the influence of different linkers and head groups on their characteristics. In a previous study, among the tested linkers (Gly, L-Ser, and β-Ala), the polar linker L-Ser enhanced probe solubility and membrane immersion and favored a desirable cholesterol tilt angle. In this study, we present our findings on five new CND analogs incorporating the L-Lys linker. This linker was selected for its unique structural features: its carboxyl group anchors it to cholesterol, the ε-amino group provides an attachment point for the naphthalimide fluorophore, and the alpha-amino group remains free and ionizable under physiological conditions. In this study, we aimed to evaluate the influence of the free alpha-amino group on the probe’s biophysical properties and compare its performance to that of the previously studied hydroxyl group from the L-serine linker. The fluorescence of the 1,8-naphthalimide probe is influenced by the nature of the substituent at the C-4 position, known as the “head group”. This moiety also plays a crucial role in determining the probe’s biophysical properties (
Figure 1).
2. Materials and Methods
All reagents and solvents (including anhydrous ones) were obtained from commercial sources (Fisher Scientific, Waltham, MA, USA) and used without purification unless stated otherwise. The LysoView™ 650 was purchased from Biotium (Fremont, CA, USA), and TopFluor-Cholesterol (TFC) was purchased from Avanti Polar Lipids (Alabaster, AL, USA).
2.1. Thin-Layer and Column Chromatography
Thin-layer chromatography (TLC) was performed on aluminum-backed plates coated with silica gel (60 Å pore diameter, 200 μm layer thickness; SiliCycle (Quebec, QC, Canada) or EMD Millipore (Merck Millipore, Burlington, VT, USA). All TLC plates contained a fluorescent indicator and were visualized under UV light (λ_ex = 254 nm or 366 nm) or by staining with a solution of 10% H2SO4 in ethanol followed by brief heating with a heat gun. Column chromatography was conducted using columns packed with silica (60 Å pore diameter, 40–60 μm particle size; SiliCycle).
2.2. NMR and Mass Spectrometry
NMR spectra were recorded on 400 MHz spectrometer at 22 °C in d-chloroform or d4-methanol. Proton (1H) and carbon (13C) signals were referenced to tetramethylsilane (TMS), and signal multiplicities are denoted as singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), multiplet (m), broad (br), or combinations thereof. Mass spectra were obtained using a Bruker Microflex MALDI-TOF spectrometer (Bruker Corporation, Billerca, MA, USA) with α-cyano-4-hydroxycinnamic acid (α-CHCA) as the matrix and were recorded in positive ion mode.
2.3. Synthesis of CND Probes
The synthesis scheme and detailed methods for preparing the CND probes are provided in the
Supplementary Material.
2.4. Determination of Absorbance Spectra in Organic Solvents
Stock solutions of the CND probes were diluted to 10 μM in chloroform and evaporated to dryness. The residues were re-dissolved in dichloromethane, dimethyl sulfoxide, ethanol, or hexane to yield 10 μM solutions. Absorbance spectra were recorded from 300 to 700 nm using a Thermo Scientific Evolution 201 UV–VIS spectrophotometer (Thermo Scientific, Waltham, MA, USA).
2.5. Determination of Emission Spectra in Organic Solvents
Stock solutions of the CND probes were diluted to 1 μM in chloroform and evaporated to dryness. The residues were re-dissolved in the respective solvents to yield 1 μM solutions. Emission spectra were recorded using a fixed excitation wavelength of 400 nm on a PerkinElmer LS 55 fluorescence spectrometer (PerkinElmer, Waltham, MA, USA), scanning from 420 to 650 nm.
2.6. Determination of Molar Extinction Coefficients in DMSO and CHCl3
Stock solutions of the CND probes were prepared at 100 µM in the respective solvents. For each data point, the solutions were serially diluted by a factor of 0.75 in triplicate. Absorbance values were recorded at the wavelength corresponding to the absorbance maximum for each solvent using a Thermo Scientific Evolution 201 UV–VIS spectrophotometer.
2.7. Fluorescence Dependence on pH in 1% Octyl-β-Glucoside (OG) Solution
OG solutions (1%) were prepared at pH values ranging from 5 to 10 using Sorenson’s phosphate buffer. After gentle mixing, the solutions were allowed to equilibrate for 30 min. A stock solution of the CND probes in DMSO was diluted to 20 μM. For each pH, 95 μL of the OG solution was added to a well in a 100 μL well plate, followed by 5 μL of dye solution, with the solution aspirated three times. The plate was then incubated in the darkness for 15 min. Fluorescence measurements were conducted using a Spectramax Gemini EM plate reader (Molecular Devices, San Jose, CA, USA) with a fixed excitation wavelength of 405 nm, scanning the visible range with an excitation cut-off at 455 nm. Fluorescence values at the emission maximum were plotted against pH.
2.8. Preparation and Analysis of Lipid Phase Partitioning in Giant Unilamellar Vesicles (GUVs)
Lipid stock solutions of DOPC (10 mM), brain sphingomyelin (10 mM), cholesterol (10 mM), DiD (100 μM), and CND dye (40 μM) were prepared in a 9:1 CHCl
3:MeOH mixture, and the GUVs were formed in water containing 50 mM sucrose as described previously [
11] using the Vesicle Prep Pro (VPP) instrument (Nanion Technologies, Munich, Germany). GUV images were acquired on a Nikon A1R confocal microscope (Nikon, Melville, USA) with a 20× objective. CND detection was performed with a 405 nm excitation and an emission range of 500–550 nm, while DiD was excited at 640 nm with an emission range of 663–738 nm. Individual vesicle images were cropped from the raw data, and vesicles displaying clear phase separation in the DiD channel were analyzed using the GUV-AP plugin to quantify liquid-ordered (Lo) partitioning [
12]. Vesicle identification and stitching were performed using the DiD channel with a threshold coefficient of 0.12, a circle extension coefficient of 1.2, and an angle step of 3°. The fraction of probes present in the Lo phase was calculated according to the following equation:
where FLo represents the average fluorescence intensity of the circular segment corresponding to the Lo phase, and FLd represents the average fluorescence intensity of the circular segment corresponding to the Ld phase.
2.9. Molecular Dynamics Simulations and Analysis
CND probe molecules were constructed using the CHARM-GUI server’s input generator, ligand reader, and modeler modules [
13,
14,
15]. A positive charge was added to the piperazine group as well as α-amino group of lysine to represent its protonation state at physiological pH. CHARMM-compatible topology and parameter files were generated using CGenFF parametrization protocol [
16]. Membrane structures were built using the membrane builder, positioning the probe such that the naphthalimide moiety resided at the membrane surface and cholesterol was embedded within the hydrophobic core [
17]. The membrane model consisted of 25% sphingomyelin (SSM), 25% cholesterol, and 50% POPC, with lipids symmetrically distributed between leaflets. Neutral pH and physiological salt conditions (0.15 M NaCl) were maintained, and the systems were charge neutral. Molecular dynamics simulations were carried out using the CHARMM36m force field and the GROMACS 2022.1 package with GPU acceleration [
18]. Following system construction, energy minimization was performed using the steepest descent method with hydrogen bond constraints using the LINCS algorithm [
19]. The system underwent six equilibration phases at 298.15 K as per the CHARMM-GUI protocol, followed by a 200 ns production run in the NPT ensemble using the Nose–Hoover thermostat. Electrostatics were calculated using the particle mesh Ewald method with a 1.2 nm cut-off. The final 150 ns of each trajectory were used for analysis. Trajectory data were processed in Visual Molecular Dynamics (VMD, ver. 1.9.4) [
20], and membrane properties such as cholesterol tilt and thickness were evaluated using the MembPlugin [
21]. Bilayer thickness was determined from the positions of phosphorus atoms, while cholesterol tilt angles were calculated by analyzing the positions of the C10 and C13 carbons relative to the membrane normal defined by the phosphorus atom mass distribution. Immersion values were determined by calculating the distance of selected oxygen atoms (the C-3 bound oxygen and, where applicable, the hydroxyl oxygen) from the membrane center, defined as the average of the phosphorus atom positions in each leaflet. Hydrogen bonds between the probe and membrane components (POPC, sphingomyelin, water) were quantified using VMD’s H-bond plugin with a donor-acceptor distance cut-off of 3 Å and an angle cut-off of 30°. These analyses were performed on every frame of the final 150 ns of the trajectories. A total hydrogen bond count is reported for all CNDs. For cholesterol, the hydrogen bonds are presented as an average that is calculated by dividing the total number of bonds by the number of cholesterol molecules, providing a per molecule value. Representative CND structures were obtained with clustering analysis using UCSF Chimera (ver. 1.16) [
22], with coordinates extracted from the top ensemble representative frame.
2.10. Cell Culture
NIH-3T3 fibroblasts were obtained from ATCC (ATCC, Manassas, VA, USA). A vial of cryopreserved cells was thawed in a 37 °C water bath and immediately plated in 75 mL culture flasks pre-coated with Matrigel (Corning, Corning, NY, USA). Cells were cultured to confluence in DMEM supplemented with GlutaMax and 5% fetal bovine serum. Confluent cultures were washed with PBS and detached using 1% Trypsin/EDTA. Following centrifugation, the cell pellets were resuspended in the same media and plated onto quad-divided glass-bottom 35 mm culture dishes at a density of 1,000,000 cells/cm2. Cultures were maintained in the same media and used within 2–5 days.
2.11. Live-Cell Fluorescence Confocal Imaging
Live-cell imaging was performed using a Nikon A1R confocal microscope equipped with a 60× (N.A. 1.40) oil immersion objective and excitation lasers with appropriate filters for CNDs (405 nm excitation), TopFluor-Cholesterol (TFC) (488 nm excitation), and LysoView 650 (640 nm excitation). Image acquisition was carried out using Nikon Elements, with laser power, gain, and offset settings optimized to maximize dynamic range while minimizing fluorophore bleed-through; these settings were maintained consistently across experiments. Cells were incubated with the desired probe to a final concentration of 1 μM for 1 h, followed by incubation in dye-free media for the required time period. Prior to imaging, cells were loaded with 0.5 μM LysoView 650 for 1 h, then washed with dye-free media, and placed in an Oko-Lab weather chamber (Okolab, Pozzuoli, Italy) mounted on the microscope stage. The chamber and the 60× objective were pre-warmed to 37 °C, and the chamber was maintained at 5% CO
2 and 100% humidity. For each sample, three random fields of view were captured at a resolution of 1024 × 1024 pixels. Z-stack images were acquired with a step size of 0.3 μm, and time-lapse imaging was performed with a 10-s frame interval over 3–5 min. Image analysis was subsequently performed using ImageJ/Fiji (ver. 2.16.0) [
23].
2.12. Particle Analysis
Fluorescence intensity across Z-stacks was evaluated, and a representative frame (with high fluorescence intensity) was extracted for further analysis after applying the “despeckle” (median) filter. Regions of interest (ROIs) were generated using trainable Weka segmentation [
24]. The classifier was trained on series of low signal-to-noise and high signal-to-noise images. Classified images were converted to masks, and ROIs were subsequently outlined. Particle analysis was conducted with Fiji/ImageJ, using unprocessed images with object detection limited to areas between 0.1 µm
2 and 10 µm
2. For each ROI, the mean pixel intensity and area were determined, and particle diameters were estimated assuming circular geometry. Further analysis was limited to particles ranging from 0.3 to 1 µm. Data from 1-h and 24-h time points were compared and plotted using GraphPad Prism (version 10), with statistical significance assessed using the Wilcoxon matched pairs signed rank test.
2.13. Colocalization Analysis
Pearson’s correlation coefficient (PCC) and Manders’ colocalization coefficient (MCC) values [
25] were determined using the EzColocalization plugin in Fiji/ImageJ [
26]. Colocalization was performed on single frames from the Z-stack (selected based on maximum green channel intensity) with Costes’ thresholding. PCC and MCC values were obtained for each analog in triplicate. Data from 1-h and 24-h experiments were compared and plotted using GraphPad Prism (version 10), and statistical analysis was conducted using the Wilcoxon matched pairs signed rank test.
2.14. Visualization and Drawing Software
Reaction schemes and 2D structural formulas were prepared using MarvinSketch (ChemAxon Ltd., Budapest, Hungary, version 21.15,
https://www.chemaxon.com, accessed on 1 March 2025), while three-dimensional molecular visualizations were generated with UCSF Chimera (version 1.16) [
22]. Some plots were obtained using RStudio (version 2024.04).
4. Discussion
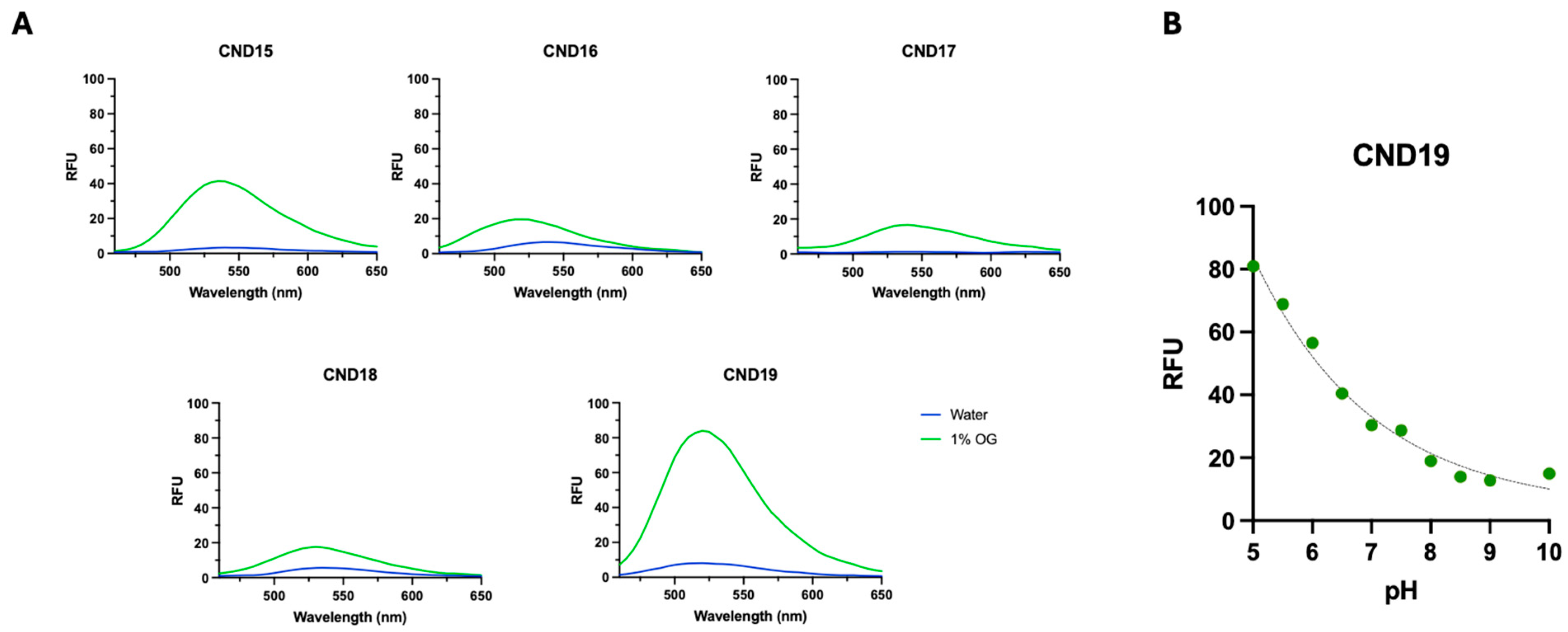
The synthesized CND15–CND19 analogs were designed to investigate how L-lysine linker and distinct head groups influence their photophysical properties, lipid interactions, and cellular uptake, with the goal of evaluating their potential as fluorescent cholesterol probes. The incorporation of a lysine linker aimed to enhance hydrophilicity and cellular uptake through its positively charged α-amino group, potentially affecting lipid ordering. Our findings demonstrate that these structural modifications significantly influence the solvatochromic behavior, membrane partitioning, molecular orientation, and intracellular localization of these probes. The solvatochromic properties of CND15–CND19 revealed strong environmental sensitivity, a characteristic of 1,8-naphthalimide fluorophores. The observed red shifts in polar solvents, particularly in DMSO, indicate the presence of a charge transfer excited state, stabilized by solvent polarity [
38]. Notably, CND15 exhibited the highest molar extinction coefficient and longest absorption wavelength, suggesting enhanced conjugation and stronger electronic transitions compared to the other analogs. In contrast, CND17 displayed the lowest extinction coefficient and a blue-shifted absorption maximum, indicative of weaker charge transfer interactions. Fluorescence measurements in octyl glucoside (OG) micelles versus water provided further insights into the probes’ aggregation tendencies. The higher fluorescence intensity of CND15, CND17, and CND19 in OG micelles suggests reduced self-quenching, whereas CND16 and CND18, which contain hydrophobic head groups, exhibited increased aggregation in aqueous environments. The pH-sensitive fluorescence of CND19 aligns with previous reports on piperazine-containing CND analogs, further confirming that the head group modulates the probe’s response to its environment [
29,
39]. Lipid phase partitioning studies using GUVs revealed distinct preferences among the analogs for liquid-ordered (Lo) and liquid-disordered (Ld) phases. CND17 and CND16 exhibited the strongest Lo phase affinity (~40%), while CND19, the most polar analog, displayed the lowest (~25%). These results indicate that both linker and head group polarity play a key role in Lo phase localization, reflecting earlier findings with cholesterol-based CND membrane probes. Furthermore, the partitioning behavior of these analogs correlates with their head group characteristics, underscoring the crucial contributions of linker hydrophobicity, hydrogen bonding capacity, and fluorophore orientation. Additionally, the positioning of the fluorophore along the linker plays a pivotal role in dictating phase partitioning preference.
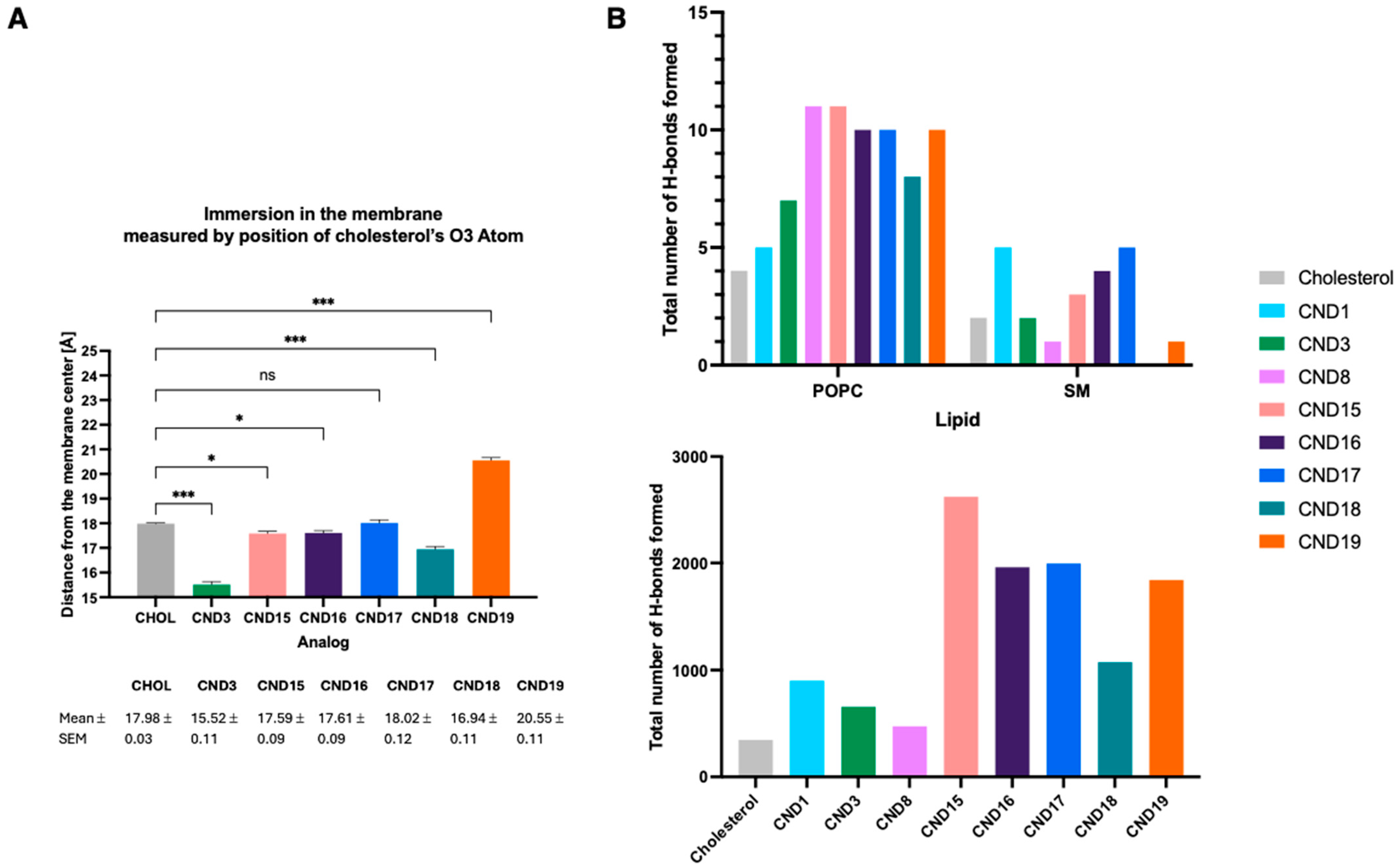
Molecular dynamics simulations provided additional mechanistic insights into the behaviors of CND15–CND19 within lipid membranes. The calculated cholesterol tilt angles indicated significant deviations from native cholesterol, with CND16 and CND18 displaying the highest tilt angles, likely due to their hydrophobic head groups promoting deeper membrane embedding. The presence of the α-amino group influenced probe positioning within the bilayer, leading to increased interactions with lipid phosphate groups and water molecules. Surprisingly, even the hydrophobic analogs CND16 and CND18 were positioned higher in the membrane than cholesterol, suggesting that electrostatic interactions with the linker play a dominant role in membrane positioning. These results emphasize that both the head group and linker chemistry collectively determine how the probe integrates into lipid membranes.
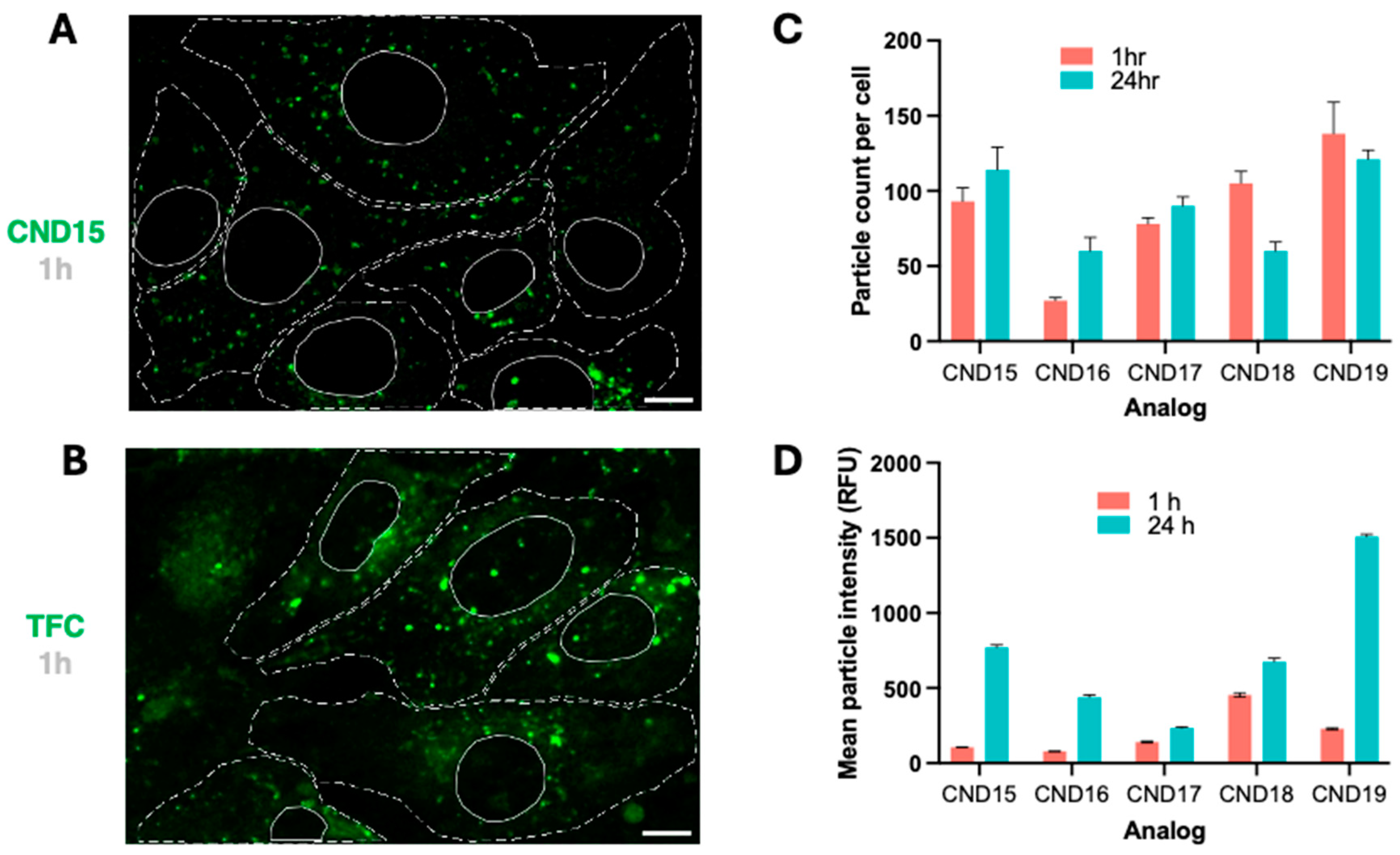
Live-cell imaging revealed that all CND analogs localized intracellularly within vesicular structures, suggesting endosomal trafficking. The uptake efficiency varied significantly among the analogs, with CND15 and CND19 exhibiting the highest uptake at 1 h post-incubation, whereas CND16, with its hydrophobic piperidine head group, displayed the lowest uptake. Interestingly, CND18, containing a morpholine moiety, was taken up more efficiently than CND16, likely due to its slightly higher polarity. We have observed the same phenomenon with CND6 which was extremely well taken up by 3T3 fibroblasts, but not astrocytes. These findings highlight the impact of head group charge/polarity on cellular uptake efficiency.
Over 24 h, the number of intracellular CND particles remained stable or slightly increased, except for CND18, which exhibited a decline in fluorescence. This behavior mirrors that of CND6, which shares the same head group, suggesting a common mechanism influencing intracellular retention. Analysis of fluorescence intensity changes over time confirmed that all analogs accumulated in vesicles, with the extent of accumulation being head group dependent. CND19, featuring a piperazine head group, exhibited the most pronounced increase in fluorescence intensity, followed by ethanolamine-functionalized CND15. In contrast, CND17, with its hydroxyl head group, displayed the smallest increase. Notably, at 24 h, CND19 produced the strongest fluorescence signal, twice as intense as CND15, a trend also evident at 1 h. This enhanced fluorescence in CND19 is attributed to the pH-sensitive piperazine moiety, which, upon protonation inside the acidic lysosomes, inhibits photoinduced electron transfer (PET), thereby enhancing fluorescence.
Lysosomes play a crucial role in cholesterol metabolism and trafficking within cells [
40]. Within the lysosome, acid lipases hydrolyze cholesterol esters to release free cholesterol [
41]. This free cholesterol is then exported from the lysosome through the coordinated action of proteins like NPC1 and NPC2, allowing it to be redistributed to other cellular compartments such as the endoplasmic reticulum and plasma membrane [
40]. In this sense, lysosomes act as a central hub for both the processing and redistribution of cholesterol, helping maintain cellular cholesterol homeostasis.
The colocalization analysis using Pearson’s correlation coefficient (PCC) and Manders’ colocalization coefficient (MCC) confirmed lysosomal accumulation of CNDs over time. PCC values increased across all analogs from 1 to 24 h, indicating progressive lysosomal localization. The highest PCC values were observed for CND19, followed by CND15, whereas CND16 displayed the lowest correlation, suggesting minimal lysosomal association. MCC values consistently exceeded PCC values, confirming substantial lysosomal colocalization. At 24 h, MCC values surpassed 0.7 for all analogs, with CND15 and CND19 showing the highest lysosomal retention. The progressive increase in both PCC and MCC values suggests that lysosomes play a key role in CND trafficking, with piperazine-containing CND19 exhibiting the strongest lysosomal association.
The performance of CND15–CND19 was compared to previously reported CND1-10 probes [
11]. Although CND15 and CND19 exhibited best fluorescent signal intensity and cellular uptake among CND15–CND19 analogs, their preference for Lo partitioning was less optimal compared to CND3, which features a serine linker with a hydroxyl group at the beta position. These results suggest that the presence of an amino group in the alpha position of lysine introduces strain in the linker, affecting the interactions of the head group attached to the naphthalimide fluorophore. Consequently, this constrain leads to diminished Lo partitioning compared to probes featuring a serine linker. On the other hand, lysine-based analogs bearing a positively charged amino group accumulate in cells far more effectively than those bearing glycine or serine linkers published earlier [
11]. We hypothesize that this enhanced accumulation arises from the increased positive charge, which promotes stronger interactions with phospholipids. These findings underscore the critical role of linker design in modulating the probe performance.
CND probes constitute a novel class of fluorescent cholesteryl probes built on a 1,8-naphthalimide (ND) scaffold, offering a modular design that allows flexible variation of linkers and head groups to fine-tune their properties. Compared with established probes such as NBD-cholesterol and BODIPY-modified cholesterol (Bchol) [
6], CND probes feature substantially larger Stokes shifts (~130 nm vs. ~40 nm for NBD-cholesterol and ~10 nm for Bchol), making them particularly advantageous for co-imaging with other fluorescent reporters. In model membranes (GUVs), CND probes exhibit Lo/Ld partitioning ranging from 30 to 50%, with CND3 closely matching Bchol’s partitioning behavior. Notably, CND probes generally show stronger Lo domain partitioning than 22- or 25-NBD analogs. Furthermore, several members of this series (CND2–CND4, CND19) display pH-dependent fluorescence via a piperazine head group, providing additional versatility for imaging applications.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}