Abstract
Flax (Linum usitatissimum L.), as the earliest oil and fiber crop, is a model plant for genetic inferences of plant domestication processes involving multiple domestication events. However, a puzzle has emerged from several genetic studies, as dehiscent cultivated flax is genetically more related to its progenitor pale flax (L. bienne Mill.), and winter cultivated flax is well mixed with oil or fiber cultivated flax, while capsular dehiscence and winter hardiness are the major characteristics of pale flax. For this, a comparative analysis was conducted with 16 Linum samples representing pale flax and four domestication groups of cultivated flax (oil, fiber, winter, and dehiscent) using 454 pyrosequencing, Sanger resequencing and microsatellite data. It was found that the genomic sampling of genetic variants from the three applied methods yielded similar genetic information on pale flax and four groups of cultivated flax. The revealed genetic relationships did not show significant departures from the previous findings, but instead supported an early, independent domestication of a primitive flax lineage for oil use, followed by a subsequent flax domestication process with multiple domestication events for capsular dehiscence, oil, fiber and winter hardiness. Domestication on capsular dehiscence occurred earlier than domestication on winter hardiness. Domestication on winter hardiness was more complicated than domestication on capsular dehiscence.
1. Introduction
Flax (Linum usitatissimum L.) was the first founder crop domesticated for oil and fiber uses in Near Eastern agriculture roughly 8000 years ago [1,2,3]. Genetic studies have identified pale flax as the wild progenitor of cultivated flax [4,5,6,7]. The archaeological finds of pale flax came first from Tell Abu Hureyra in northern Syria (11,200–10,500 years ago) [8] and occurred throughout the Near East by the 8th millennium BC [9]. The archaeological records from Tell Ramad in Syria (9000 years ago) revealed the first occurrence of cultivated forms of flax with an increase in seed size [2]. Flax then spread from the Near East to Europe and the Nile Valley [3]. The archaeological finds in southwest Germany revealed larger flax seeds in the early, rather than later, phase of the Late Neolithic (4000–2500 BC) [10]. The flax varieties that spread into the Danube Valley were winter oil varieties. However, summer fiber varieties developed in eastern Europe also spread into central Europe [11] and replaced the original varieties [1,12]. All modern fiber varieties in use today are thought to have originated from eastern Europe [1,11]. However, the rest of the early history of flax domestication remains unclear [9,13].
Pale flax (L. bienne Mill.), or previously L. usitatissimum L. subsp. angustifolium (Huds.) Thell. [14]), is a winter annual or perennial plant and has narrow leaves, dehiscent capsules, and large variation in the vegetative plant parts and growth habit [12,15,16]. In contrast, cultivated flax still keeps variable seed dormancy and has fast growth with large variation in the generative plant parts, early flowering, almost-indehiscent capsules and large seeds. These phenotypic differences reflect the major domestication syndromes of cultivated flax in capsular openness, seed size, oil yield, plant stem, and winter habit [12,17]. Interestingly, some domestication syndromes have been used to group cultivated flax such as dehiscent cultivated flax with spontaneously opening capsules and winter flax with a vernalization requirement [18,19,20,21]. These groups of cultivated flax with domestication syndromes should carry genetic signatures of flax domestication accumulated over time. Assessments of genetic relationships among various groups of cultivated flax with unique domestication traits [22] may shed some insight into the flax domestication paths [22,23].
Flax was cultivated for both oil and fiber production. The dual purpose of domestication makes flax a unique model for genetic inferences of complex crop domestication processes [22,23,24,25,26]. Recent molecular inferences suggest that cultivated flax probably descended from a single domestication of pale flax, apparently for its oil, rather than fiber, use [6,7,13,23]. Also, dehiscent cultivated flax is genetically more related to pale flax [22,27], and winter cultivated flax is closely related to oil or fiber cultivated flax [28]. These genetic relationships are somehow puzzling for interpreting flax domestication history, given that capsular dehiscence and winter hardiness are the major characteristics of pale flax, but winter cultivated flax is distantly related to its progenitor [22,28]. The possible explanation is that these findings were clouded with inadequate sampling of diverse flax [13,22] and/or limited genomic sampling with insufficient molecular markers [23]. For example, the most informative genetic inferences of flax domestication so far were based on the genetic signals from the sad2 locus alone [13,23], but these inferences may be biased against the domestication signals for fiber flax, as the sad2 gene is genetically more associated with oil than fiber [29].
To understand the generality of the sad2-based findings, a specific comparative analysis was conducted from 2010 to 2012 with the specific objective to assess the impacts of genomic sampling on the inferences of genetic relationship of cultivated flax and pale flax. Specifically, 16 Linum samples representing pale flax and four domestication groups of cultivated flax (oil, fiber, winter, and dehiscent) were assayed using three different genomic sampling methods. The first was the application of the most advanced method of Roche 454 pyrosequencing in 2010 [30] to sample genome-wide genetic variants for better resolution to acquire domestication signals. As the informativeness of Roche 454 pyrosequencing was not known at the initial stage of next-generation sequencing, the second method used was Sanger resequencing analysis based on some contigs generated from Roche 454 pyrosequencing to focus on specific genomic regions. The third method was to apply expressed sequence tag-derived microsatellite (or simple sequence repeat; EST-SSR) markers to sample genetic variants in the genic region of flax genome. These three employed methods represented the most common and advanced approaches available in 2010 for sampling genetic variants to infer plant genetic relationships.
This short communication was recently generated to report the findings from the comparative analysis with the aims to address two specific questions: (1) whether the genomic sampling of genetic variants from three applied methods revealed compatible patterns of genetic relationships on pale flax and four domestication groups of cultivated flax and (2) whether the revealed genetic relationships among five assayed Linum groups from these genomic approaches are compatible with the previous sad2-based findings.
2. Materials and Methods
2.1. Plant Materials and DNA Extraction
Sixteen genetically diverse Linum accessions, originating from 10 countries (Table 1), were selected for this study to represent pale flax and four domestication groups of cultivated flax with major domestication syndromes (high oil content, strong bast fiber, indehiscent capsule, and winter habit). Approximately ten seeds were randomly chosen from each selected accession maintained at Plant Gene Resources of Canada, Saskatoon, Canada. Selected seeds were planted in multi-pots filled with a regular soilless mix. Plants were grown for two to three weeks for cultivated flax and up to two months for pale flax (due to dormancy) in a greenhouse at the Saskatoon Research Centre, Agriculture and Agri-Food Canada. The greenhouse conditions were 22 °C during the day and 16 °C at night, with a photoperiod of 16 h between 4 am and 8 pm. Young leaf tissue from individual plants of each accession was collected, freeze-dried, and stored at −20 °C. For this study, one individual plant was randomly selected to represent its accession, and DNA was extracted from 15 mg of freeze-dried tissue of each selected plant using the DNeasy Plant Mini Kit (Qiagen, Mississauga, ON, Canada) following the manufacturer’s instructions, quantified using the Thermo Scientific Nanodrop 8000, and adjusted to 100 ng/µL using Qiagen AE buffer (10 mM Tris-HCl, 0.5 mM EDTA, pH 9.0) for various analyses below.

Table 1.
List of 16 studied Linum accessions representing pale flax (L. bienne) and four domestication groups of cultivated flax (L. usitatissimum).
2.2. 454. Pyrosequencing
The 454 pyrosequencing of the 16 samples and the SNP data collection were described in detail by Fu and Peterson [30]. Briefly, the DNA samples were first subjected to a genomic reduction and multiplex identifiers (MID) barcoding by digesting with EcoRI and BfaI and ligating with BfaI and biotin-modified EcoRI adaptors. Eight barcoded DNA samples were combined into one of two pools. The pooled DNA was submitted to the DNA Technologies Laboratory at the Canadian National Research Council’s Plant Biotechnology Institute (Saskatoon, SK, Canada) and sequenced using the Roche 454 GS FLX instrument with Titanium chemistry. The bioinformatics analysis of the 454 sequence reads to identify contigs and SNPs includes several major steps. First, the sequence reads were separated into sample specific SFF files based on MID barcodes using the Roche Newbler SFF tools, followed by removal of the forward and reverse adaptor sequences. Contig assembly and SNP detection were performed using the DIAL pipeline [31]. The pipeline adds the SFF file of each sample and performs a completely automatic call of contigs and SNPs from all added SFF files. Additional efforts were made with custom PERL scripts to report the assembled contigs and SNPs for further data analysis. This SNP data set was named 454-SNP data.
2.3. Sanger Resequencing
Sanger resequencing was performed on 24 contigs that were generated from the 454 pyrosequencing, and these selected contigs had the most putative SNPs, representing the most polymorphic loci. The procedure for validating the selected contigs with three samples was the same as described in Fu and Peterson [30], and the same primer sets used for contig validation were applied (Table S1). Forward and reverse Sanger sequences from each sample were assembled using Sequencher v.4.10.1 (GeneCodes, Ann Arbor, MI, USA), aligned using Muscle v.3.6 [32] against the consensus sequence of each contig, and proofread by hand. All the Sanger sequences were aligned with the contig consensus sequences. The putative SNPs were manually checked with the available Sanger sequences, and additional SNPs and indels were also identified, if any, from the Sanger resequencing. The resulting SNP data set was named Sanger-SNP data.
2.4. EST-SSR Analysis
An EST-SSR analysis of the 16 samples was performed with 19 of the most informative EST-SSR primer pairs. These primer pairs were developed by Cloutier et al. [33] and characterized by Fu and Peterson [34] and Fu [27] (see Table S2). The analysis followed the same experimental procedures of Fu and Peterson [34], including PCR conditions, PCR protocol, visualization of PCR products, and scoring of DNA fragments. This analysis generated an EST-SSR data set.
2.5. Data Analysis
Levels of polymorphism were first analyzed for each data set. For 454-SNP data, the number of contigs detected with SNPs, the total numbers of SNPs observed, and the number of missing sequence reads were calculated for each sample. For Sanger-SNP data, the length of the assayed contig, the number of SNPs predicted on the contigs, the number of SNPs confirmed with the 16 samples, the number of new SNPs and indels, and the total number of SNPs and indels were calculated for each contig. These measurements were also calculated for all the sequences concatenated for all 16 samples. The nucleotide diversity was also estimated using DnaSP program [35] on concatenated sequences for five Linum groups. For EST-SSR data, the number of polymorphic bands per locus and the related summary statistics on band frequencies were generated for all 16 samples. The diversity content per locus was estimated with Shannon entropy following Russell et al. [36], as the entropy does not require strict genetic assumptions. This entropy-based diversity content essentially measures the effective number of alleles per marker locus [37]. These analyses were conducted with a SAS program written in SAS IML [38].
For each data set, genetic relationships of the 16 Linum accessions were obtained using three commonly applied approaches. First, the genetic relationships were inferred using PAUP* [39] with a neighbor-joining method, and a radiation tree was displayed using MEGA v.4.01 [40]. However, this tree may be confounded with recombination and lineage sorting. Second, a distance-based NeighborNet [41] of the 16 samples was generated using the SplitsTree4 [42] with the options of Uncorrected_P and EqualAngle. This tree displayed detailed reticulations where recombination or lineage sorting may occur. Third, the maximum clade credibility phylogenies were generated using BEAST v.1.4 [43] with a relaxed uncorrelated lognormal clock and with tree prior as constant size, expansion, or exponential growth. The HYK substitution model was used with gamma distribution for site heterogeneity for DNA sequence data, and under simple model for binary with microsatellite data. The other options were used with default values. The Bayesian Markov chain Monte Carlo approach implemented in BEAST should reveal more informative relationships with evolutionary rates from sequence data carrying recombination signals, as it directly estimates ultrametric phylogenies and model parameters and takes into account both the branch length errors and the topological uncertainties [44].
The optimal genetic structure of the 16 samples was also inferred with the model-based Bayesian method available in the BAPS software [45]. The individual samples were clustered using the model for non-linked markers and 20 replicate runs of the algorithm were performed with the upper-bound values (K) from 2 to 10 for the number of clusters. This was performed for each data set.
An analysis of molecular variance was performed using Arlequin v.3.01 [46] to assess genetic variation within and among various Linum groups. The group-specific proportion of genetic variation (Fst) allows for a measure of genetic variation within a Linum group. A higher Fst value means less genetic diversity within the group. The significance of variance components and related genetic distances was tested with 10,000 random permutations. This analysis was repeated for each data set.
3. Results
The 454 pyrosequencing generated 1067 SNPs scored from 450 of the 713 identified contigs for the 16 samples. However, the 454-SNP data were highly unbalanced, as each sample had an unequal number of sequence reads with variable read quality. The percentage of SNP data missing for a sample ranged from 42.2% (atalanteO) to 97.8% (turkey3P) and averaged 68.9%. The cultivated flax samples had 162 species-specific SNPs, while only 3 species-specific SNPs were observed in the pale flax samples. The Sanger resequencing generated a total of 6886 bp of concatenated sequences on the 24 contigs, confirmed 165 (88.2%) predicted SNPs, and detected 119 new SNPs and 19 indels (Table S1). In total, these 24 contigs had 284 putative SNPs among 16 samples and effective sequence lengths ranging from 131 to 441 base pairs per contig. There were 16 contigs with gene annotations from the first flax genome assembly and annotation [47], mainly with different proteins (Table S1). Screening 19 EST-SSR primer pairs identified a total of 157 polymorphic bands across the 16 samples (Table S2). The number of bands detected per primer pair ranged from 2 to 12 with an average of 8.3 bands per primer pair. An assessment of diversity content per primer pair revealed a relatively high Shannon entropy ranging from 0.62 to 3.24 and averaging 2.3 (Table S2).
The maximum clade credibility (MCC) trees of the 16 Linum samples with respect to data set were shown in Figure 1. For the 454-SNP data, the pale flax samples were clearly separated by their country origin. The dehiscent flax samples were closely related to the pale flax samples, rather than to other cultivated flax samples. The winter flax samples were clustered with oil and fiber flax samples (Figure 1A). The similar patterns of genetic relationships were observed for the EST-SSR data, with slightly higher resolutions with the dehiscent flax departing from the pale flax samples (Figure 1B). However, the Sanger-SNP based MCC tree displayed more variation for the dehiscent and winter flax samples (Figure 1C). The winter flax sample from Egypt and dehiscent flax sample from Russia were closely related to one pale flax sample from Turkey (Figure 1C). Similarly, the dehiscent flax samples from Portugal and Turkey were closely related to another pale flax sample from Turkey. The winter flax samples from Israel and Turkey were closely related to the oil cultivar labeled atalanteO. For all the data sets, the oil flax samples were always slightly more related to the pale flax samples than the fiber flax samples, which is consistent with the sad2-based findings [13,23]
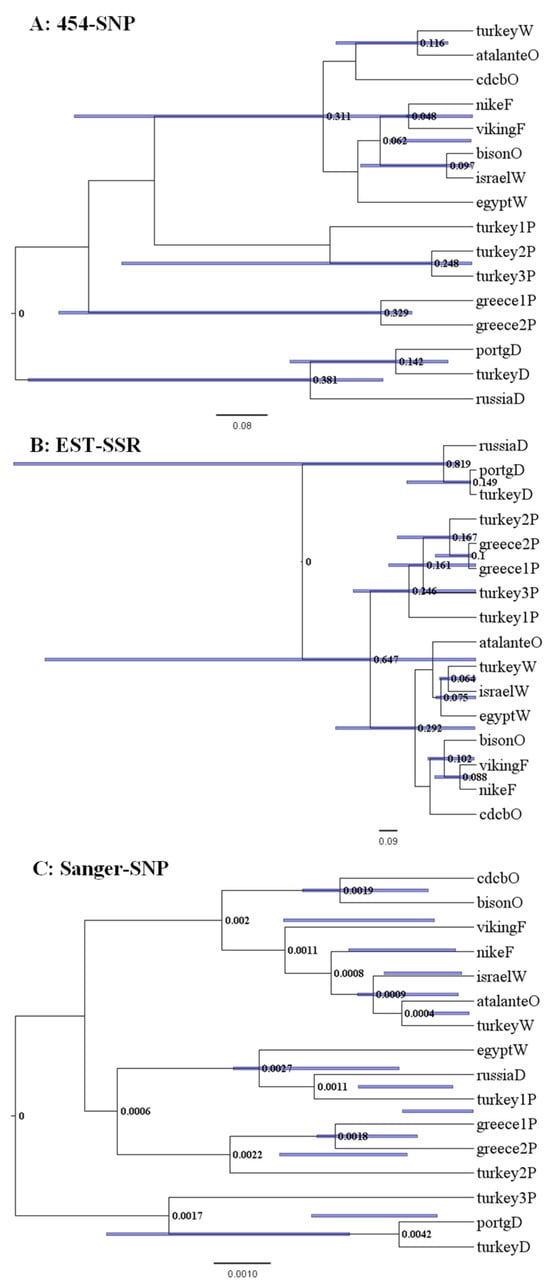
Figure 1.
The maximum clade credibility trees of 16 Linum accessions representing pale flax and four domestication groups of cultivated flax revealed by the BEAST program based on three data sets ((A): 454-SNP, (B): EST-SSR, (C): Sanger-SNP). The node length and node bar for Length_95%_HPD are shown. The last letter of the sample label represents the flax group (Table 1). Note that each tree has its own scale of estimated distance.
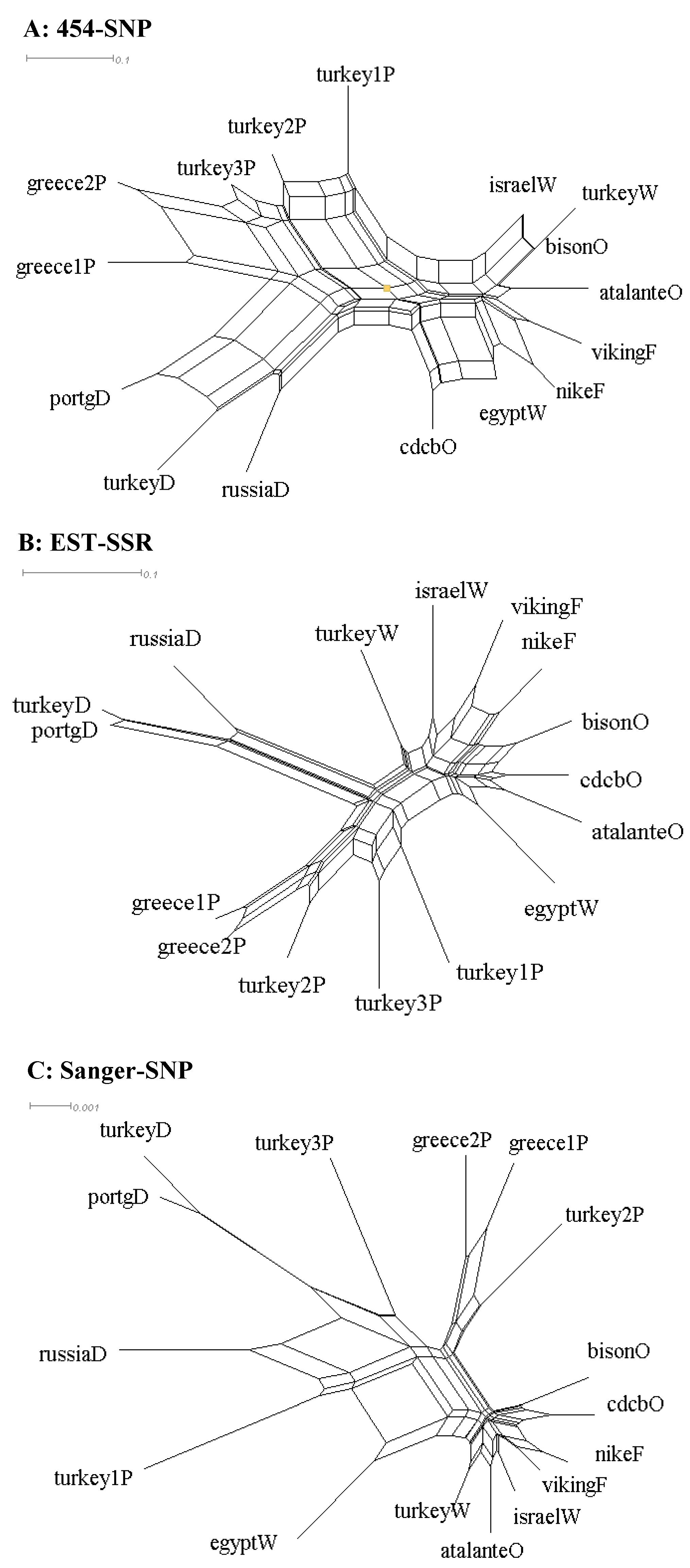
The same patterns of genetic relationships for these 16 samples were observed based on the NeighborNet derived from the SplitsTree4 program (Figure 2). First, the NeighborNet clearly mirrored the MCC trees for each data set, but the former displayed reticulation information. For example, roughly half of the branch length for most of the branches in the NeighborNet with the Sanger-SNP data was confounded with the reticulations. Second, the largest reticulation was observed in the NeighborNet from the 454-SNP data, followed by that from the EST-SSR data. Also, the MCC trees were further supported by the neighbor-joining trees of the 16 samples inferred using PAUP* for all the data sets. The topologies were the same with the MCC trees with respect to data set, but branch lengths varied for most branches (results not shown). It is worth noting that for ease of interpretation, the comparative analyses of Linum genetic relationships were conducted with consideration of the MCC tree or NeighborNet topologies only, not the variations of branch length (even shown) in each MCC tree (Figure 1) or NeighborNet (Figure 2).
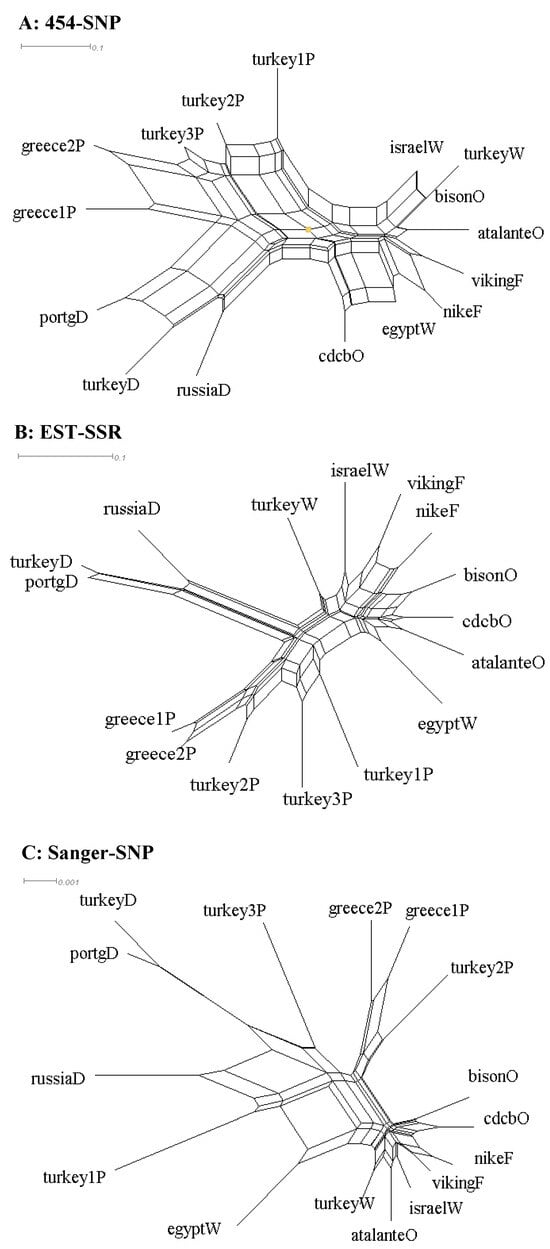
Figure 2.
The NeighborNets of 16 Linum accessions representing pale flax and four domestication groups of cultivated flax revealed by the SplitsTree4 program based on three data sets ((A): 454-SNP, (B): EST-SSR, (C): Sanger-SNP). The last letter of the sample label represents the flax group (Table 1). Note that each NeighborNet has its own scale of estimated distance.
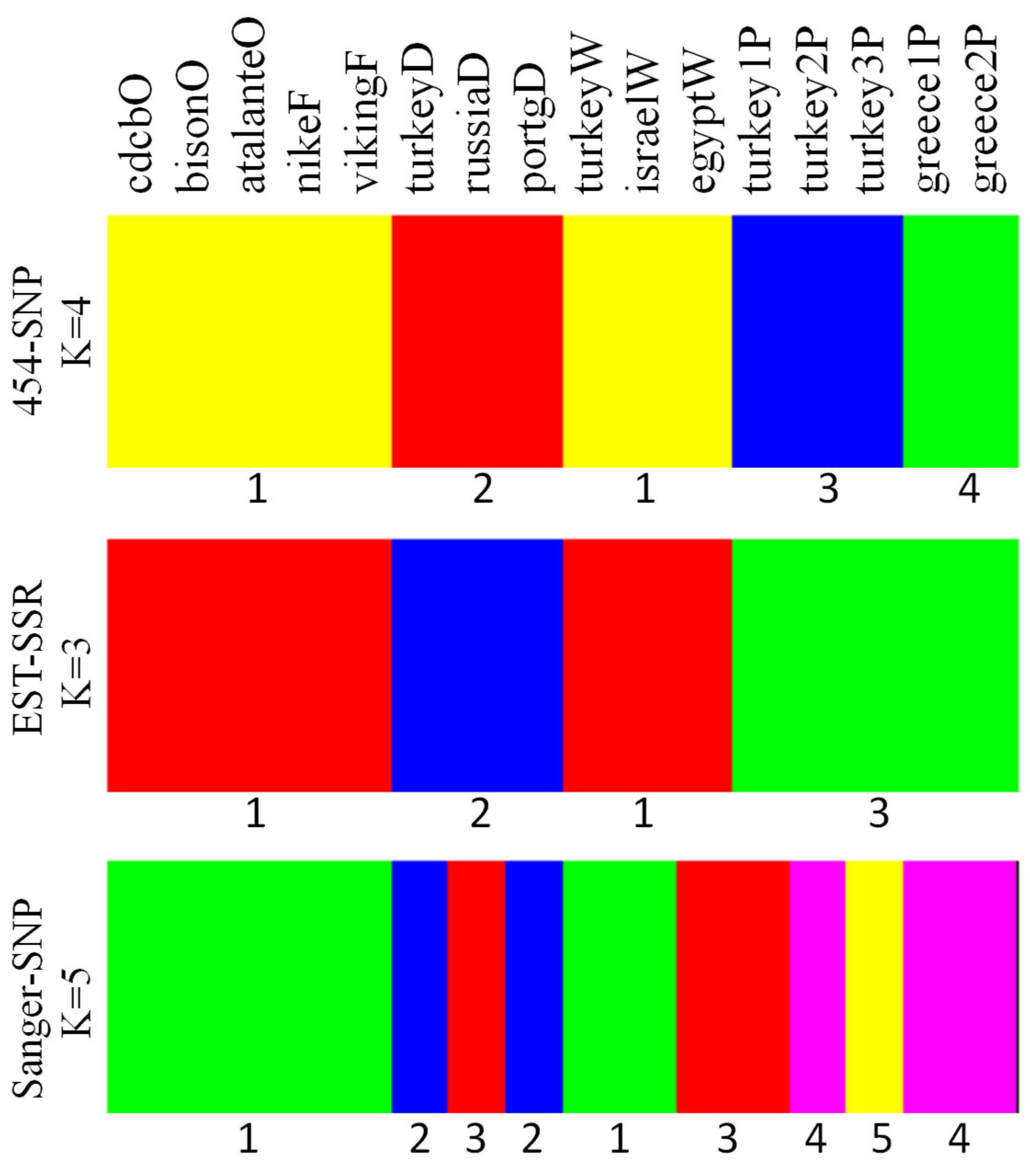
The model-based analysis of genetic structure via BAPS showed four optimal clusters with the 454-SNP data, three clusters with the EST-SSR data, and five clusters with the Sanger-SNP data (Figure 3), with the highest log likelihoods of −3263.3, −1239.2, and −3999.7, respectively. For the 454-SNP data, there were two clusters for either species. The dehiscent group was singled out from the cultivated flax, while the pale flax samples were separated by country of origin. However, the EST-SSR data grouped all the pale flax samples as one cluster and maintained the distinction of the dehiscent group from the other cultivated flax groups. In contrast to the 454-SNP and EST-SSR data, the Sanger-SNP data displayed the five pale flax samples that were spread into three clusters, one of which was mixed with one dehiscent sample (russiaD) and one winter sample (egyptW) of cultivated flax (Figure 3).
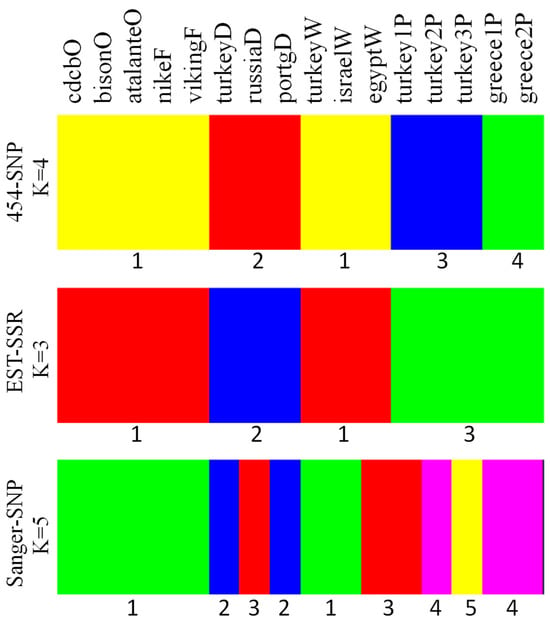
Figure 3.
Genetic clustering of 16 Linum accessions representing pale flax and four domestication groups of cultivated flax revealed by BAPS based on three data sets (454-SNP, EST-SSR, Sanger-SNP). Four optimal clusters were observed for 454-SNP; three clusters for EST-SSR; and five clusters for Sanger-SNP. Note that the corresponding clusters (which were labeled under the panel) among the three data sets may have different color labels, and the last capital letter of the sample label represents the flax group (Table 1).
Partition of total genetic variation among five Linum groups with respect to data set revealed 42–46% of the total 454-SNP and EST-SSR variation, but only 28.2% Sanger-SNP variation, residing among five Linum groups. Measured with group-specific Fst, all five of the Linum groups displayed compatible Fst values when compared between the 454-SNP and EST-SSR data, but considerable variation in Fst values inferred from the Sanger-SNP data. For example, the range of Fst values was 0.321 to 0.575 for 454-SNP, 0.345 to 0.503 for EST-SSR, and 0.141 to 0.498 for Sanger-SNP (Table 2). It remains unknown why there were such differences in group-specific Fst among three data sets. However, the group-specific Fst values for Sanger-SNP seemed to be consistent with the early reports of genetic variations present among Linum groups (e.g., [28,48]). For example, the highest Fst value of 0.498 for cultivated fiber flax (Table 2) matched well with the expectation of the lower genetic variation among the assayed groups [48]. The pale flax had Fst values ranging from 0.141 with the Sanger-SNP to 0.418 with the 454-SNP. Similarly, dehiscent and winter cultivated flax also displayed considerably large changes in Fst value ranging from 0.239 to 0.484 and 0.327 to 0.575, respectively. Considering the average pairwise nucleotide diversity with the Sanger sequence data alone, the pale flax samples showed the highest nucleotide diversity of 0.01002, followed by the dehiscent group (0.00830) and winter group (0.00628) (Table 2).
Table 2.
Comparisons of proportional genetic variation among pale flax and four groups of cultivated flax obtained from the analysis of molecular variance based on three data sets.
The proportional marker variation present in a pair of Linum groups ranged from 0.116 to 0.592 and averaged 0.405 for 454-SNP; from 0.156 to 0.650 and 0.407 for EST-SSR; and from 0.080 to 0.526 and 0.260 for Sanger-SNP (Table 2). On the average, the dehiscent group had the largest differentiations with the other four groups for any data set, but these differentiations were not statistically significant at p < 0.05, except with the pale flax. Interestingly, the pale flax group had significant or nearly significant differentiations with the four groups of cultivated flax, consistently for all three data sets.
4. Discussion
The results presented in this short communication were generated more than 12 years ago, but they are still useful for our understanding of flax domestication processes in three aspects. First, the genomic sampling of genetic variants from three applied methods yielded similar genetic information on pale flax and four domestication groups of cultivated flax. Second, the results confirmed the previous findings generated from limited genetic markers [22,27,28], even at one locus [23], that dehiscent cultivated flax is genetically more related to pale flax than the other three groups of cultivated flax [22,27] and that winter cultivated flax is more closely related to oil or fiber cultivated flax than pale flax [28]. Third, the revealed genetic relationships further confirmed that the domestication of flax on winter hardiness was more complicated than domestication on capsular dehiscence [27,28].
The three data sets collected for this comparison carried genetic signals from different parts of the flax genome and these genetic signals should differ from those at the sad2 locus [23]. The 454-SNPs sampled genome-wide genetic variants, Sanger-SNPs surveyed genetic variants at 24 specific regions with 16 associated with functional genes, and EST-SSR acquired genetic signals from genic regions of the flax genome. However, these data sets also had limits in their signal resolutions. For example, the 454-SNP data was highly unbalanced for the 16 assayed samples with missing SNP data, the Sanger-SNP data sampled only 24 chromosomal segments, and the EST-SSR data were generated from only 19 primer pairs. With the limited resolution, it is difficult to determine which data set provided the most informative inferences of Linum genetic relationships, although the original configuration of this comparative analysis favored 454-SNP data [30]. Also, it is not surprising to observe some inconsistencies of the revealed genetic relationships among the three data sets. For example, the Sanger-SNP data revealed that the winter cultivated flax egyptW and the dehiscent cultivated flax russiaD were closely related to the pale flax turkey1P (Figure 1C), but such genetic relationships were not observed in the 454-SNP data (Figure 1A) and EST-SSR data (Figure 1B). Also, the comparison was based on 16 Linum samples only and the coverage of flax genetic diversity was low, clearly showing the weakness of this comparative analysis. Ideally, each assayed group is required to have 30 or more samples with better geographic and genetic diversity coverages to make more informative comparison of the employed genomic sampling methods in the inferences of flax domestication processes. These limitations can be easily resolved now with whole-genome sequencing [49] and expanded sample size for better resolution, but the comparison was made in 2012 at the initial stage of next generation sequencing. However, further expanded research using advanced tools of next generation sequencing is in progress.
Flax is a model plant for genetic inferences of plant domestication processes with multiple domestication events [13,23]. With the advances in genomic tools (e.g., [50]), the inferences can provide better insights into flax domestication origin in the Near East and its spread into northern Europe, like the other domestication plants of wheat (e.g, [51]), rice (e.g., [52]), and maize (e.g., [53]). However, the major challenge is the lack of pale flax germplasm collected across its species distribution range. At the time, there were only 114 pale flax accessions with GPS coordinates conserved in several seed gene banks [16], which is not sufficient for genetic inferences of flax domestication with better resolution. Thus, efforts are needed to collect pale flax germplasm across its species distribution range, not only for flax domestication studies, but also for flax germplasm conservation [16].
The revealed genetic relationships of these Linum samples have some implications for our understanding of flax domestication. These genetic relationships supported the previous findings that there was an early, independent domestication of a primitive flax lineage for oil use, followed by a subsequent flax domestication process with multiple domestication events for capsular dehiscence, oil, fiber and winter hardiness. Domestication on capsular dehiscence occurred earlier than domestication on winter hardiness and seemed to be simpler than domestication on winter hardiness. These results could be explained, as the need to improve winter tolerance was realized only when flax spread from the Near East into Europe [3,11,54,55]. Also, selection for capsular dehiscence would have been more efficient than selection for winter hardiness, as capsular dehiscence is controlled by fewer genes than winter hardiness which is expected to be governed by many genes of small effects [56]. However, the most interesting confirmation is the variable genetic relationships of winter cultivated flax with oil, fiber and pale flax [23,28], which is different from dehiscent cultivated flax [27]. This implies a difficulty of dating and describing the domestication for winter hardiness. Domestication for winter hardiness could be a gradual process toward the higher latitudes of Europe [11], as the flax spread over Europe was involved with both oil and fiber flax [1,3,54,55]. To facilitate further research on flax domestication, we posed a hypothesis that winter cultivated flax experienced differential domestication pressure over the flax spread into Europe, and searching for ancestral genomic diversity [57] with respect to flax winter hardiness can recover the domestication processes for winter hardiness.
5. Conclusions
This comparative analysis revealed that three genomic samplings of genetic variants yielded similar genetic information on pale flax and four domestication groups of cultivated flax. The revealed genetic relationships supported the previous findings that there was an early, independent domestication of a primitive flax lineage for oil use, followed by a subsequent flax domestication process with multiple domestication events for capsular dehiscence, oil, fiber and winter hardiness. Domestication on capsular dehiscence occurred earlier than domestication on winter hardiness. Domestication on winter hardiness was more complicated than domestication on capsular dehiscence.
Supplementary Materials
The following supporting information can be found online at Figshare DOI (https://doi.org/10.6084/m9.figshare.25955029 (accessed on 29 May 2024)): Table S1. List of 24 primer pairs used for Sanger resequencing of 24 contigs in 16 Linum samples, along with the polymorphism and gene annotation information and Table S2. List of 19 EST-SSR primer pairs used in this study to genotype 16 Linum accessions and their polymorphisms detected.
Funding
This research was funded by AAFC research grants J-000066, J-000185 and J-003159 to Yong-Bi Fu.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Some supplemental data can be found in the Supplementary Materials.
Acknowledgments
The author would like to thank Greg Peterson and Carolee Horbach for their technical support in 454 pyrosequencing, Sanger resequencing, EST-SSR screening and scoring of Linum samples; Robin Allaby, Jeff Ross-Ibarra and Benjamin Kilian for their useful discussions on this research; and Carolee Horbach for her assistance in the preparation of the manuscript and three anonymous journal reviewers for their helpful comments on the early version of this manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Helbaek, H. Domestication of food plants in the Old World. Science 1959, 130, 365–372. [Google Scholar] [CrossRef]
- Van Zeist, W.; Bakker-Heeres, J.A.H. Evidence for linseed cultivation before 6000 BC. J. Archaeol. Sci. 1975, 2, 215–219. [Google Scholar] [CrossRef]
- Maier, U.; Schlichtherle, H. Flax cultivation and textile production in Neolithic wetland settlements on Lake Constance and in Upper Swabia (south-west Germany). Veg. Hist. Archaeobot. 2011, 20, 567–578. [Google Scholar] [CrossRef]
- Tammes, T. The genetics of the genus Linum. Bibliogr. Genet. 1928, 4, 1–36. [Google Scholar]
- Gill, K.S. Linseed; Indian Council of Agricultural Research: New Delhi, India, 1987. [Google Scholar]
- Fu, Y.B.; Peterson, G.; Diederichsen, A.; Richards, K.W. RAPD analysis of genetic relationships of seven flax species in the genus Linum L. Genet. Resour. Crop Evol. 2002, 49, 253–259. [Google Scholar] [CrossRef]
- Fu, Y.B.; Allaby, R.G. Phylogenetic network of Linum species as revealed by non-coding chloroplast DNA sequences. Genet. Resour. Crop Evol. 2010, 57, 667–677. [Google Scholar] [CrossRef]
- Hillman, G. The plant remains from Tell Abu Hureyra: A preliminary report. Proc. Prehist. Soc. 1975, 41, 70–73. [Google Scholar]
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin; Oxford University Press: New York, NY, USA, 2012; pp. 100–113. [Google Scholar]
- Herbig, C.; Maier, U. Flax for oil or fiber? Morphometric analysis of flax seeds and new aspects of flax cultivation in Late Neolithic wetland settlements in southwest Germany. Veg. Hist. Archaeobot. 2011, 20, 527–533. [Google Scholar] [CrossRef]
- Gutaker, R.; Zaidem, M.; Fu, Y.B.; Diederichsen, A.; Smith, O.; Ware, R.; Allaby, R. Flax latitudinal adaptation at LuTFL1 altered architecture and promoted fiber production. Sci. Rep. 2019, 9, 976. [Google Scholar] [CrossRef]
- Diederichsen, A.; Hammer, K. Variation of cultivated flax (Linum usitatissimum L. subsp. usitatissimum) and its wild progenitor pale flax (subsp. angustifolium (Huds.) Thell.). Genet. Resour. Crop Evol. 1995, 42, 263–272. [Google Scholar] [CrossRef]
- Allaby, R.G.; Peterson, G.W.; Merriwether, D.A.; Fu, Y.B. Evidence of the domestication history of flax (Linum usitatissimum) from genetic diversity of the sad2 locus. Theor. Appl. Genet. 2005, 112, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Hammer, K. Linaceae. In Verzeichnis Landwirtschaftlicher und Gärtnerischer Kulturpflanzen; Mansfeld, R., Schultze-Motel, J., Benedix, E.H., Fritsch, R., Grebenscikov, I., Eds.; Akademie-Verlag: Berlin, Germany, 1986; pp. 710–713. [Google Scholar]
- Uysal, H.; Kurt, O.; Fu, Y.B.; Diederichsen, A.; Kusters, P. Variation in phenotypic characters of pale flax (Linum bienne Mill.) from Turkey. Genet. Resour. Crop Evol. 2011, 59, 19–30. [Google Scholar] [CrossRef]
- Fu, Y.B. Pale flax (Linum bienne): An underexplored flax wild relative. In The Flax Genome, Compendium of Plant Genomes; You, F.M., Fofana, B., Eds.; Springer: Cham, Switzerland, 2023; pp. 37–53. [Google Scholar]
- Hammer, K. Das Domestikationssyndrom. Kulturpflanze 1984, 32, 11–34. [Google Scholar] [CrossRef]
- Elladi, V.N. Linum usitatissimum (L.) Vav. consp. nov.—Len. In Kul’turnaja Flora SSSR, Prjadil’nye [Flora of Cultivated Plants of the USSR, Fiber Plants]; Vul’f, E.V., Vavilov, N.I., Eds.; Sel’chozgiz: Moscow, Russia; Leningrad, Russia, 1940; Volume 5, Part 1; pp. 109–207. (In Russian) [Google Scholar]
- Dillman, A.C. USDA Technical Bulletin No. 1054: Classification of Flax Varieties, 1946; United States Department of Agriculture: Washington, DC, USA, 1953.
- Kulpa, W.; Danert, S. Zur systematik von Linum usitatissimum L. Kulturpflanze 1962, 3, 341–388. [Google Scholar]
- Diederichsen, A.; Fu, Y.B. Phenotypic and molecular (RAPD) differentiation of four infraspecific groups of cultivated flax (Linum usitatissimum L. subsp. usitatissimum). Genet. Resour. Crop Evol. 2006, 53, 77–90. [Google Scholar] [CrossRef]
- Uysal, H.; Fu, Y.B.; Kurt, O.; Peterson, G.W.; Diederichsen, A.; Kusters, P. Genetic diversity of cultivated flax (Linum usitatissimum L.) and its wild progenitor pale flax (Linum bienne Mill.) as revealed by ISSR markers. Genet. Resour. Crop Evol. 2010, 57, 1109–1119. [Google Scholar] [CrossRef]
- Fu, Y.B.; Diederichsen, A.; Allaby, R.G. Locus-specific view of flax domestication history. Ecol. Evol. 2012, 2, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Zeder, M.A.; Bradley, D.G.; Emshwiller, E.; Smith, B.D. Documenting Domestication: New Genetic and Archaeological Paradigms; University of California Press: Berkeley, CA, USA; Los Angeles, CA, USA, 2006. [Google Scholar]
- Brown, T.A.; Jones, M.K.; Powell, W.; Allaby, R.G. The complex origins of domesticated crops in the Fertile Crescent. Trends Ecol. Evol. 2008, 24, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Allaby, R. Integrating the processes in the evolutionary systems of domestication. J. Exp. Bot. 2010, 61, 935–944. [Google Scholar] [CrossRef]
- Fu, Y.B. Genetic evidence for early flax domestication with capsular dehiscence. Genet. Resour. Crop Evol. 2011, 58, 1119–1128. [Google Scholar] [CrossRef]
- Fu, Y.B. Population-based resequencing revealed an ancestral winter group of cultivated flax: Implication for flax domestication processes. Ecol. Evol. 2012, 2, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Ohlrogge, J.B.; Jaworski, J.B. Regulation of fatty acid synthesis. Ann. Rev. Plant Biol. 1997, 48, 109–136. [Google Scholar] [CrossRef]
- Fu, Y.B.; Peterson, G.W. Developing genomic resources in two Linum species via 454 pyrosequencing and genomic reduction. Mol. Ecol. Resour. 2012, 12, 492–500. [Google Scholar] [CrossRef]
- Ratan, A.; Zhang, Y.; Hayes, V.M.; Schuster, S.C.; Miller, W. Calling SNPs without a reference sequence. BMC Bioinform. 2010, 11, 130. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Cloutier, S.; Niu, X.; Datla, R.; Duguid, S. Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor. Appl. Genet. 2009, 119, 53–63. [Google Scholar] [CrossRef]
- Fu, Y.B.; Peterson, G.W. Characterization of expressed sequence tag-derived simple sequence repeat markers for 17 Linum species. Botany 2010, 88, 537–543. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.R.; Hosein, F.; Johnson, E.; Waugh, R.; Powell, W. Genetic differentiation of cocoa (Theobroma cacao L.) populations revealed by RAPD analysis. Mol. Ecol. 1993, 2, 89–97. [Google Scholar] [CrossRef]
- Reyes-Valdés, M.H.; Williams, C.G. An entropy-based measure of founder informativeness. Genet. Res. 2005, 85, 81–88. [Google Scholar] [CrossRef]
- SAS Institute Inc. The SAS System for Windows V8.02; SAS Institute Inc.: Cary, NC, USA, 2004. [Google Scholar]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Moulton, V. NeighborNet: An agglomerative algorithm for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Rutschmann, F. Molecular dating of phylogenetic trees: A brief review of current methods that estimate divergence times. Divers. Distrib. 2006, 12, 35–48. [Google Scholar] [CrossRef]
- Corander, J.; Marttinen, P.; Sirén, J.; Tang, J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform. 2008, 9, 539. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Diederichsen, A.; Richards, K.W.; Peterson, G. Genetic diversity within a range of cultivars and landraces of flax (Linum usitatissimum L.) as revealed by RAPDs. Genet. Resour. Crop Evol. 2002, 49, 167–174. [Google Scholar] [CrossRef]
- You, F.M.; Moumen, I.; Khan, N.; Cloutier, S. Reference genome sequence of flax. In The Flax Genome. Compendium of Plant Genomes; You, F.M., Fofana, B., Eds.; Springer: Cham, Switzerland, 2023. [Google Scholar] [CrossRef]
- Geleta, M.; Ortiz, R. Molecular and genomic tools provide insights on crop domestication and evolution. Adv. Agron. 2016, 135, 181–223. [Google Scholar]
- Levy, A.A.; Feldman, M. Evolution and origin of bread wheat. Plant Cell 2022, 34, 2549–2567. [Google Scholar] [CrossRef] [PubMed]
- Gutaker, R.M.; Groen, S.C.; Bellis, E.S.; Choi, J.Y.; Pires, I.S.; Bocinsky, R.K.; Slayton, E.R.; Wilkins, O.; Castillo, C.C.; Negrão, S.; et al. Genomic history and ecology of the geographic spread of rice. Nat. Plants 2020, 6, 492–502. [Google Scholar] [CrossRef]
- Yang, N.; Wang, Y.; Liu, X.; Jin, M.; Vallebueno-Estrada, M.; Calfee, E.; Chen, L.; Dilkes, B.; Gui, S.; Fan, X.; et al. Two teosintes made modern maize. Science 2023, 382, eadg8940. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M. Cultivation and processing of Linum usitatissimum and Camelina sativa in southern Scandinavia during the Roman Iron Age. Veg. Hist. Archaeobot. 2013, 22, 509–520. [Google Scholar] [CrossRef]
- Karg, S.; Diederichsen, A.; Jeppson, S. Discussing flax domestication in Europe using biometric measurements on recent and archaeological flax seeds—A pilot study. In First Textiles: The Beginnings of Textile Manufacture in Europe and the Mediterranean; Siennicka, M., Rahmstorf, L., Ulanowska, A., Eds.; Oxbow Books: Oxford, UK, 2018; pp. 31–38. [Google Scholar]
- Omran, A.O.; Atkins, I.M.; Gilmore, E.C., Jr. Heritability of cold hardness in flax (Linum usitatissinum L.). Crop Sci. 1968, 8, 716–719. [Google Scholar] [CrossRef]
- Charlesworth, D. Don’t forget the ancestral polymorphisms. Heredity 2010, 105, 509–510. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Crown Copyright: © His Majesty the King in Right of Canada, 2024. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).