
Intensified Selection, Elevated Mutations, and Reduced Adaptation Potential in Wild Barley in Response to 28 Years of Global Warming




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and DNA Sequencing
2.2. SNP Calling and Annotation
2.3. Analyses of Nucleotide Diversity and Selective Sweep
2.4. Analyses of Deleterious Mutations and Mutation Burden
2.5. Analysis of Adaptation Potential
2.6. Gene Ontology and Expression Analyses
3. Results
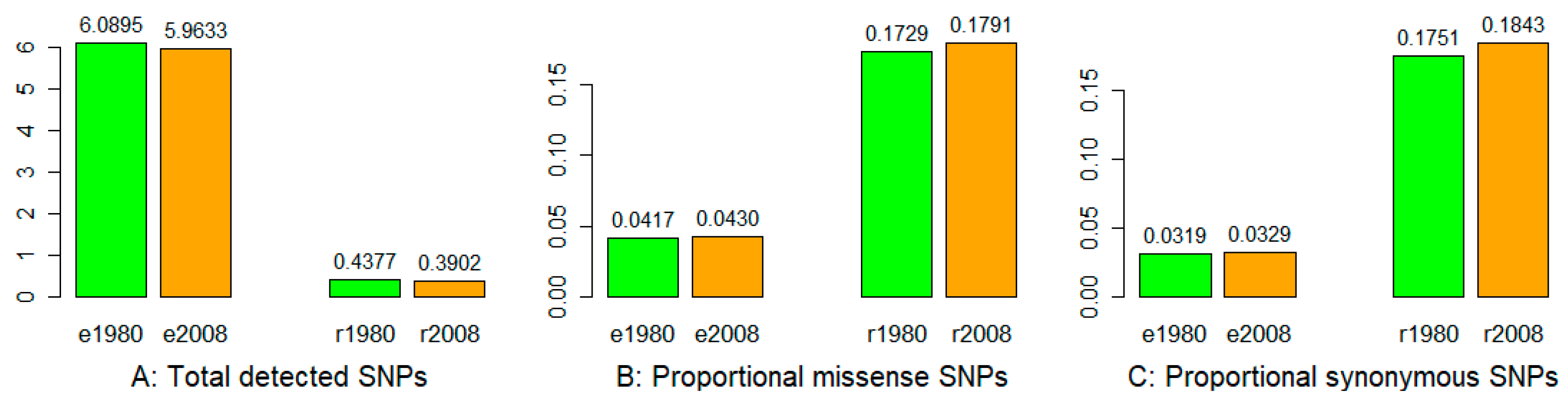
3.1. Sequencing, SNP Identification, and Annotation
3.2. Nucleotide Diversity
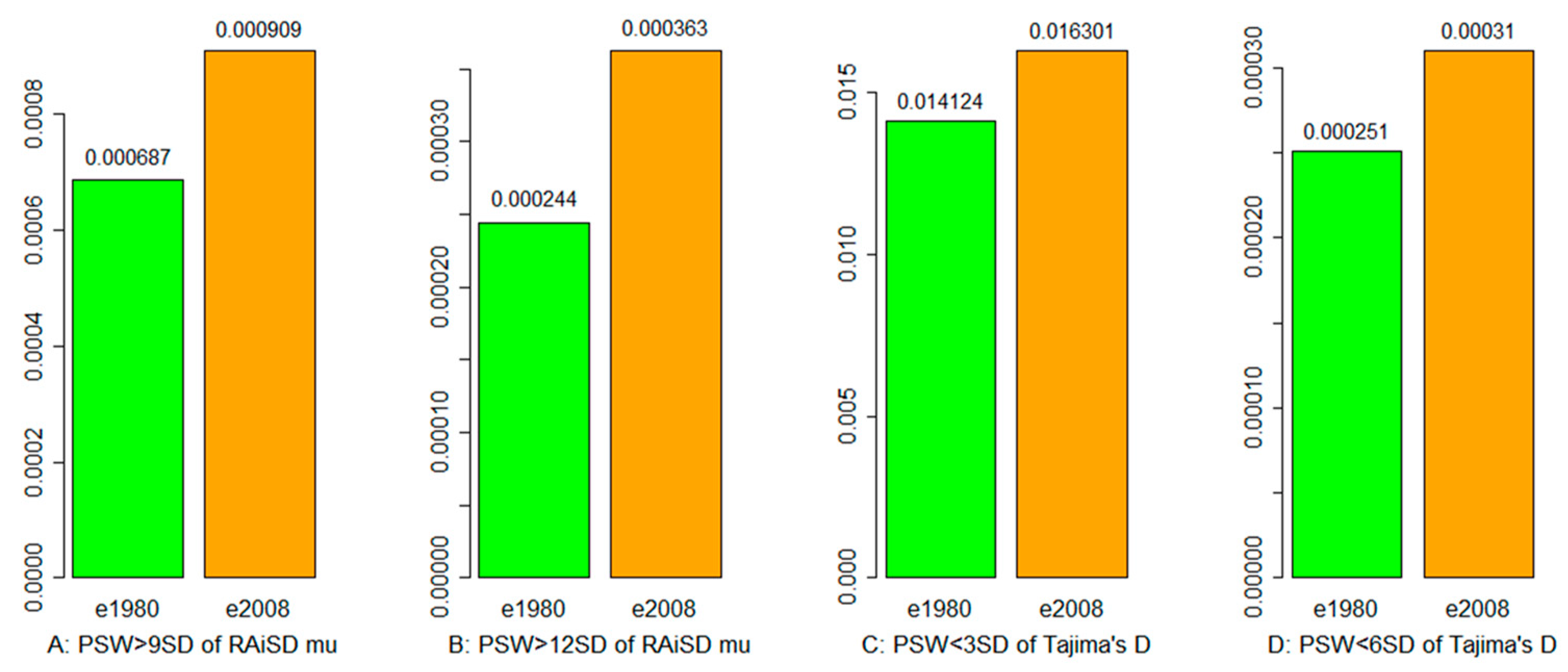
3.3. Selective Sweep
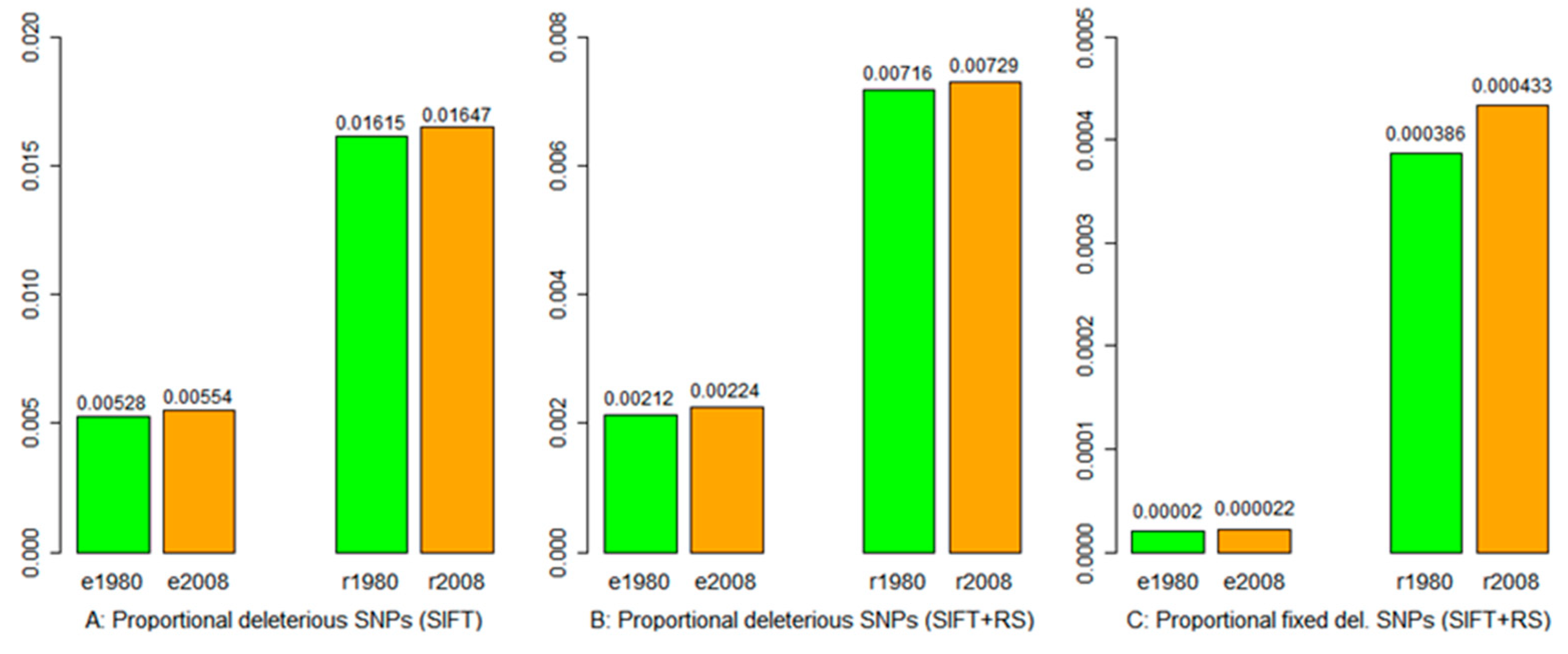
3.4. Deleterious Mutation
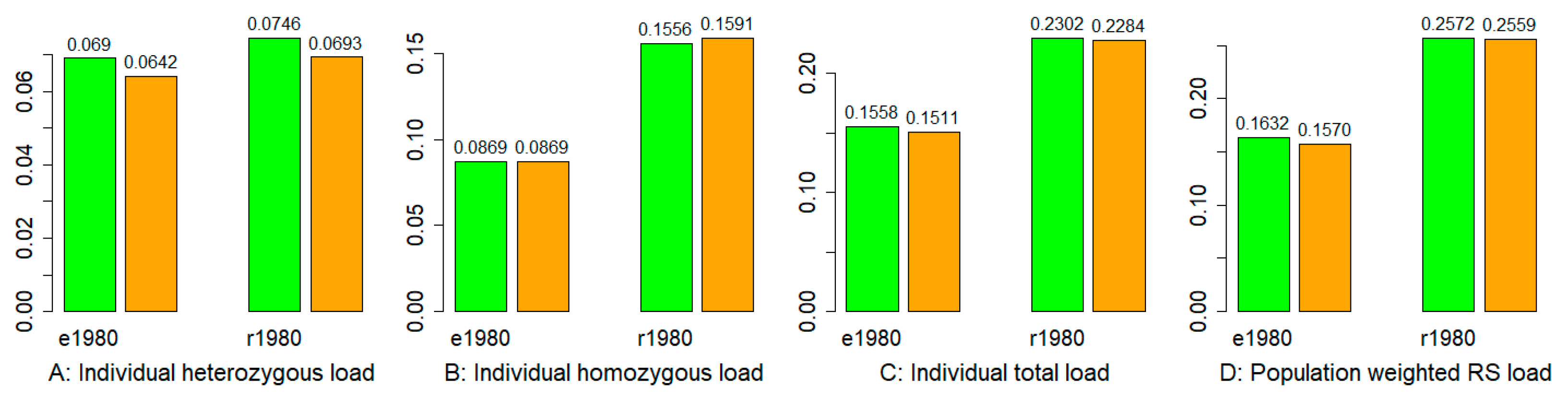
3.5. Mutation Burden
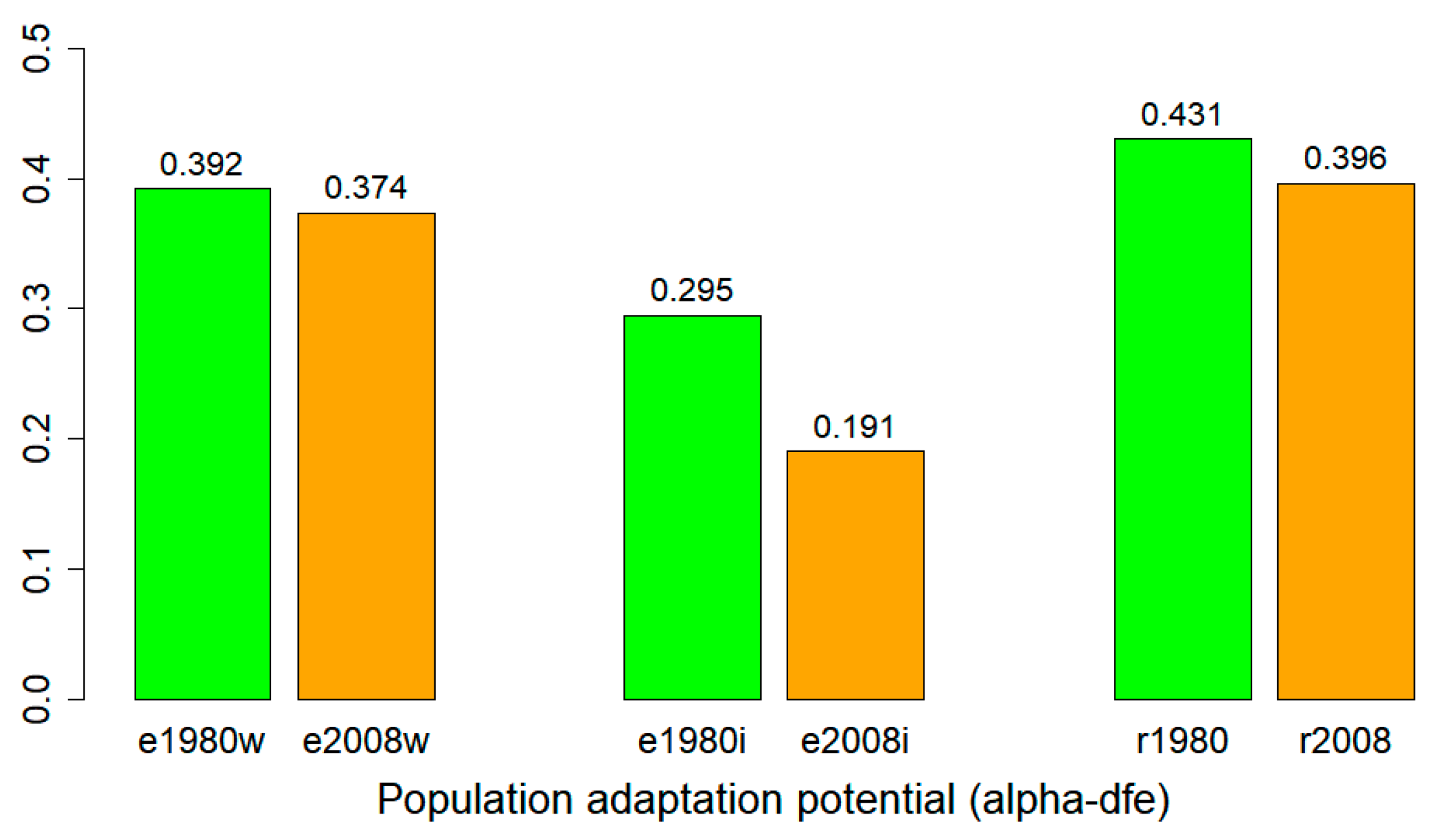
3.6. Adaptation Potential
3.7. Gene Ontology Analysis
3.8. Gene Expression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef]
- Krugman, T.; Nevo, E.; Beharav, A.; Sela, H.; Fahima, T. The Institute of Evolution Wild Cereal Gene Bank at the University of Haifa. Isr. J. Plant Sci. 2018, 65, 129–146. [Google Scholar] [CrossRef]
- Nevo, E. Origin, evolution, population genetics and resources for breeding of wild barley, Hordeum spontaneum, in the Fertile Crescent. In Barley: Genetics, Molecular Biology and Biotechnology; Shewry, P., Ed.; C.A.B. International: Wallingford, UK, 1992; pp. 19–43. ISBN 978-0-85198-725-5. [Google Scholar]
- Hajjar, R.; Hodgkin, T. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 2007, 156, 1–13. [Google Scholar] [CrossRef]
- Saranga, Y. A century of wheat research—From wild emmer discovery to genome analysis. Isr. J. Plant Sci. 2007, 55, 3–4. [Google Scholar]
- Fu, Y.B.; Somers, D.J. Genome-wide reduction of genetic diversity in wheat breeding. Crop Sci. 2009, 49, 161–168. [Google Scholar] [CrossRef]
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate trends and global crop production since 1980. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Khoury, C.K.; Amariles, D.; Soto, J.S.; Diaz, M.V.; Sotelo, S.; Sosa, C.C.; Ramírez-Villegas, J.; Achicanoy, H.A.; Velásquez-Tibatá, J.; Guarino, L.; et al. Comprehensiveness of conservation of useful wild plants: An operational indicator for biodiversity and sustainable development targets. Ecol. Indic. 2019, 98, 420–429. [Google Scholar] [CrossRef]
- Castañeda-Álvarez, N.P.; Khoury, C.K.; Achicanoy, H.A.; Bernau, V.; Dempewolf, H.; Eastwood, R.J.; Guarino, L.; Harker, R.H.; Jarvis, A.; Maxted, N.; et al. Global conservation priorities for crop wild relatives. Nat. Plants 2016, 2, 16022. [Google Scholar] [CrossRef]
- Root, T.L.; Price, J.T.; Hall, K.R.; Schneider, S.H.; Rosenzweig, C.; Pounds, J.A. Fingerprints of global warming on wild animals and plants. Nature 2003, 421, 57–60. [Google Scholar] [CrossRef]
- Thuiller, T.; Lavorel, S.; Araújo, M.B.; Sykes, M.T.; Prentice, I.C. Climate change threats to plant diversity in Europe. Proc. Natl. Acad. Sci. USA 2005, 102, 8245–8250. [Google Scholar] [CrossRef]
- Chen, I.-C.; Hill, J.K.; Ohlemuller, R.; Roy, D.B.; Thomas, C.D. Rapid range shifts of species associated with high levels of climate warming. Science 2011, 333, 1024–1026. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.; Lane, A.; Hijmans, R.J. The effect of climate change on crop wild relatives. Agric. Ecosyst. Environ. 2008, 126, 13–23. [Google Scholar] [CrossRef]
- Nevo, E.; Fu, Y.B.; Pavlicek, T.; Khalifa, S.; Tavasi, M.; Beiles, A. Evolution of wild cereals during 28 years of global warming in Israel. Proc. Natl. Acad. Sci. USA 2012, 109, 3412–3415. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Peterson, G.W.; Horbach, C.; Konkin, D.J.; Beiles, A.; Nevo, E. Elevated mutation and selection in wild emmer wheat in response to 28 years of global warming. Proc. Natl. Acad. Sci. USA 2019, 116, 20002–20008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, R.; Nevo, E.; Fu, X.; Wang, X.; Wang, Y.; Wang, C.; Chen, J.; Sun, G.; Sun, D.; et al. Genomic evidence for climate-linked diversity loss and increased vulnerability of wild barley spanning 28 years of climate warming. Sci. Total Environ. 2024, 913, 169679. [Google Scholar] [CrossRef] [PubMed]
- Parmesan, C. Ecological and evolutionary responses to recent climate change. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 637–669. [Google Scholar] [CrossRef]
- Saisho, D.; Takeda, K. Barley: Emergence as a new research material of crop science. Plant Cell Physiol. 2011, 52, 724–727. [Google Scholar] [CrossRef]
- Morin, R.; Bainbridge, M.; Fejes, A.; Hirst, M.; Krzywinski, M.; Pugh, T.; McDonald, H.; Varhol, R.; Jones, S.; Marra, M. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTechniques 2008, 45, 81–94. [Google Scholar] [CrossRef]
- Mascher, M.; Richmond, T.A.; Gerhardt, D.J.; Himmelbach, A.; Clissold, L.; Sampath, D.; Ayling, S.; Steuernagel, B.; Pfeifer, M.; D’Ascenzo, M.; et al. Barley whole exome capture: A tool for genomic research in the genus Hordeum and beyond. Plant J. 2013, 76, 494–505. [Google Scholar] [CrossRef]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Naithani, S.; Geniza, M.; Jaiswal, P. Variant Effect Prediction Analysis Using Resources Available at Gramene Database. In Plant Genomics Databases. Methods in Molecular Biology; van Dijk, A., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1533, pp. 279–297. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Alachiotis, N.; Pavlidis, P. RAiSD detects positive selection based on multiple signatures of a selective sweep and SNP vectors. Commun. Biol. 2018, 1, 79. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Moltke, I.; Albrechtsen, A.; Nielsen, R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinform. 2013, 14, 289. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Tataru, P.; Mollion, M.; Glémin, S.; Bataillon, T. Inference of distribution of fitness effects and proportion of adaptive substitutions from polymorphism data. Genetics 2017, 207, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Loewe, L.; Charlesworth, B.; Bartolomé, C.; Nöel, V. Estimating selection on nonsynonymous mutations. Genetics 2006, 172, 1079–1092. [Google Scholar] [CrossRef][Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Bedre, R.; Mandadi, K. GenFam: A new web application for gene family-based classification and functional enrichment analysis of plant genomes. Plant Direct. 2019, 3, e00191. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Fridman, E.; Mascher, M.; Himmelbach, A.; Schmid, K. Physical geography, isolation by distance and environmental variables shape genomic variation of wild barley (Hordeum vulgare L. ssp. spontaneum) in the Southern Levant. Heredity 2022, 128, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, T.A.; Sinervo, B.; Endler, J. Adaptive Genetic Variation in the Wild; Oxford University Press: New York, NY, USA, 2000; p. 265. [Google Scholar]
- Razgour, O.; Forester, B.; Taggart, J.B.; Bekaert, M.; Juste, J.; Ibáñez, C.; Puechmaille, S.J.; Novella-Fernandez, R.; Alberdi, A.; Manel, S. Considering adaptive genetic variation in climate change vulnerability assessment reduces species range loss projections. Proc. Natl. Acad. Sci. USA 2019, 116, 10418–10423. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F.; Blair, M.W. Genome–environment associations, an innovative tool for studying heritable evolutionary adaptation in orphan crops and wild relatives. Front. Genet. 2022, 13, 910386. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Villegas, J.; Jarvis, A.; Touval, J. Analysis of threats to South American flora and its implications for conservation. J. Nat. Conserv. 2012, 20, 337–348. [Google Scholar] [CrossRef]
- Brummitt, N.A.; Bachman, S.P.; Griffiths-Lee, J.; Lutz, M.; Moat, J.F.; Farjon, A.; Donaldson, J.S.; Hilton-Taylor, C.; Meagher, T.R.; Albuquerque, S.; et al. Green plants in the red: A baseline global assessment for the IUCN Sampled Red List Index for Plants. PLoS ONE 2015, 10, e0135152. [Google Scholar] [CrossRef] [PubMed]
- Newbold, T.; Hudson, L.N.; Hill, S.L.; Contu, S.; Lysenko, I.; Senior, R.A.; Börger, L.; Bennett, D.J.; Choimes, A.; Collen, B.; et al. Global effects of land use on local terrestrial biodiversity. Nature 2015, 520, 45–50. [Google Scholar] [CrossRef]
- Blanchette, M.; Kent, W.J.; Riemer, C.; Elnitski, L.; Smit, A.F.; Roskin, K.M.; Baertsch, R.; Rosenbloom, K.; Clawson, H.; Green, E.D.; et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004, 14, 708–715. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Hou, M.; Riemer, C. TOAST and ROAST. 2008. Available online: http://www.bx.psu.edu/~cathy/toast-roast.tmp/README.toast-roast.html (accessed on 7 March 2024).
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© His Majesty the King in Right of Canada, 2024. Submitted for possible open access publication under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.-B.; Peterson, G.W.; Nevo, E.; Badea, A. Intensified Selection, Elevated Mutations, and Reduced Adaptation Potential in Wild Barley in Response to 28 Years of Global Warming. Sci 2024, 6, 16. https://doi.org/10.3390/sci6010016
Fu Y-B, Peterson GW, Nevo E, Badea A. Intensified Selection, Elevated Mutations, and Reduced Adaptation Potential in Wild Barley in Response to 28 Years of Global Warming. Sci. 2024; 6(1):16. https://doi.org/10.3390/sci6010016
Chicago/Turabian StyleFu, Yong-Bi, Gregory W. Peterson, Eviatar Nevo, and Ana Badea. 2024. "Intensified Selection, Elevated Mutations, and Reduced Adaptation Potential in Wild Barley in Response to 28 Years of Global Warming" Sci 6, no. 1: 16. https://doi.org/10.3390/sci6010016
APA StyleFu, Y.-B., Peterson, G. W., Nevo, E., & Badea, A. (2024). Intensified Selection, Elevated Mutations, and Reduced Adaptation Potential in Wild Barley in Response to 28 Years of Global Warming. Sci, 6(1), 16. https://doi.org/10.3390/sci6010016