
Trifluoromethylated Quinolone-Hydantoin Hybrids: Synthesis and Antibacterial Evaluation

 , , and
, , and
Abstract
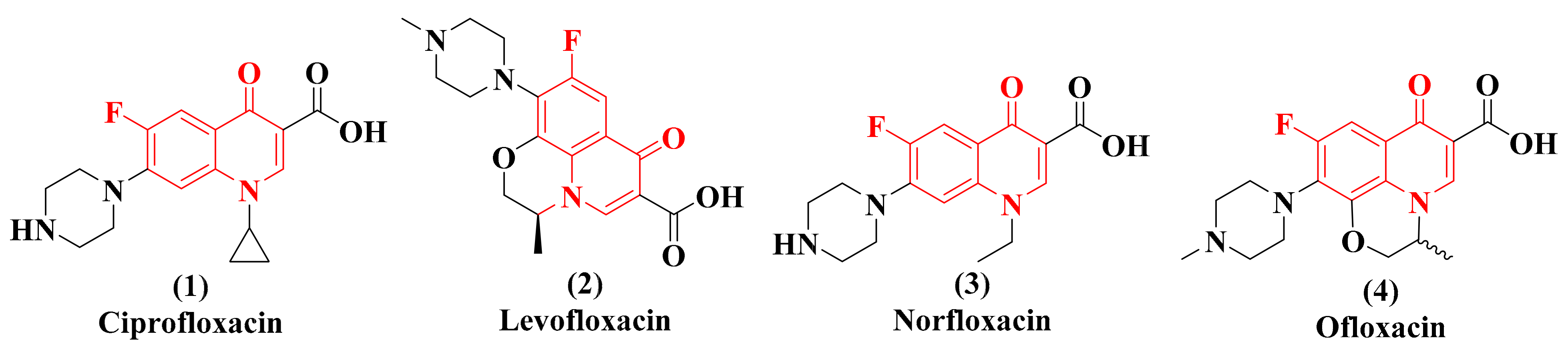
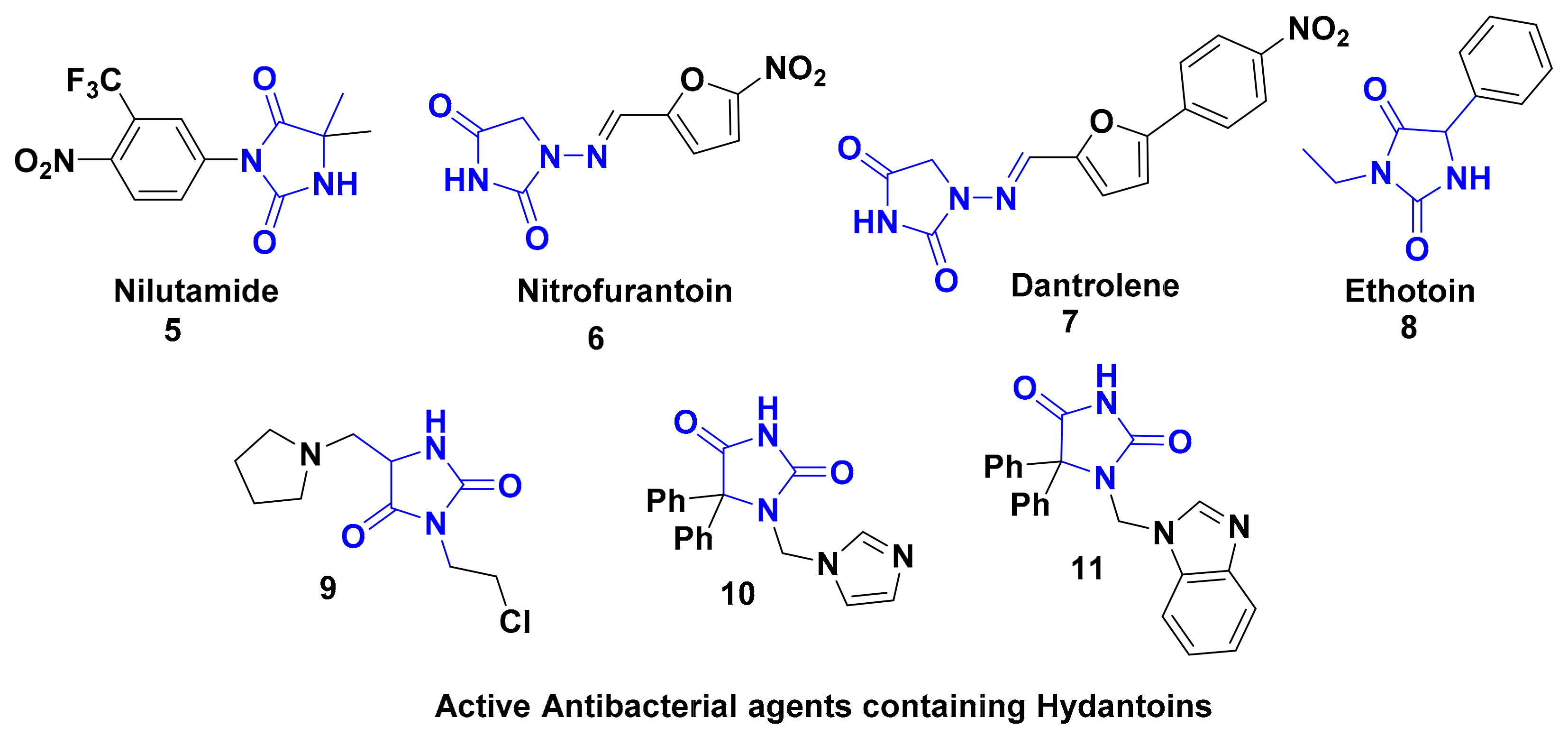
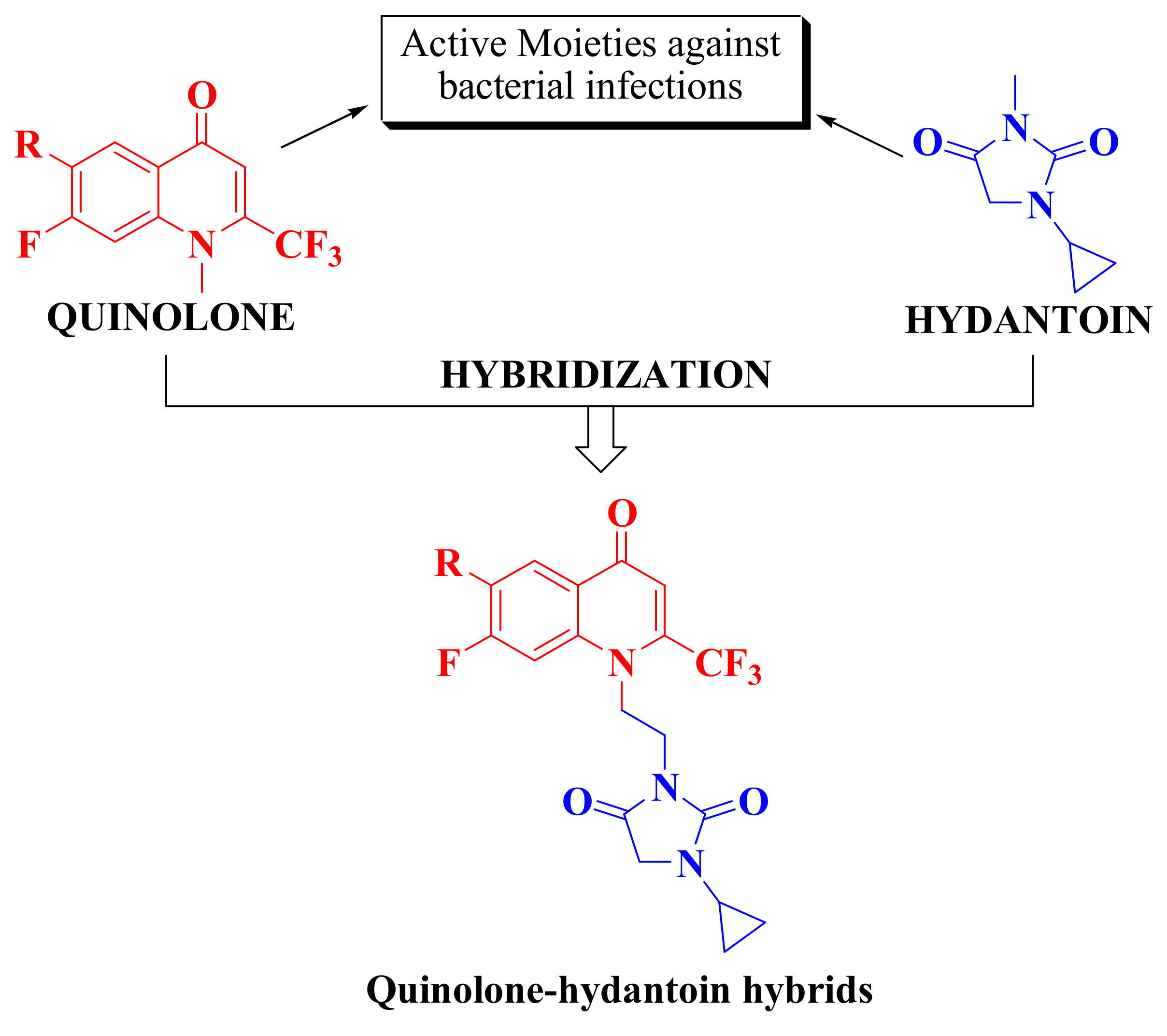
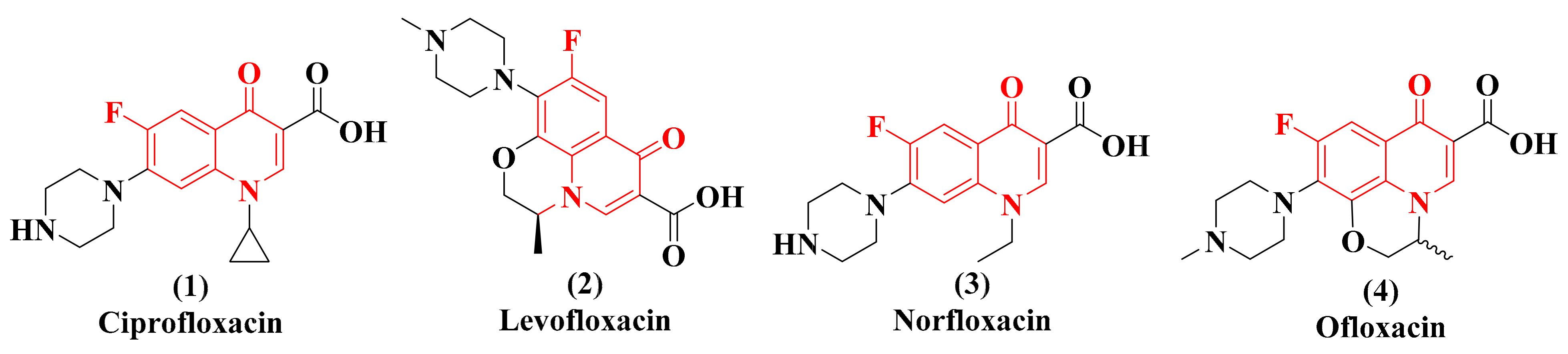
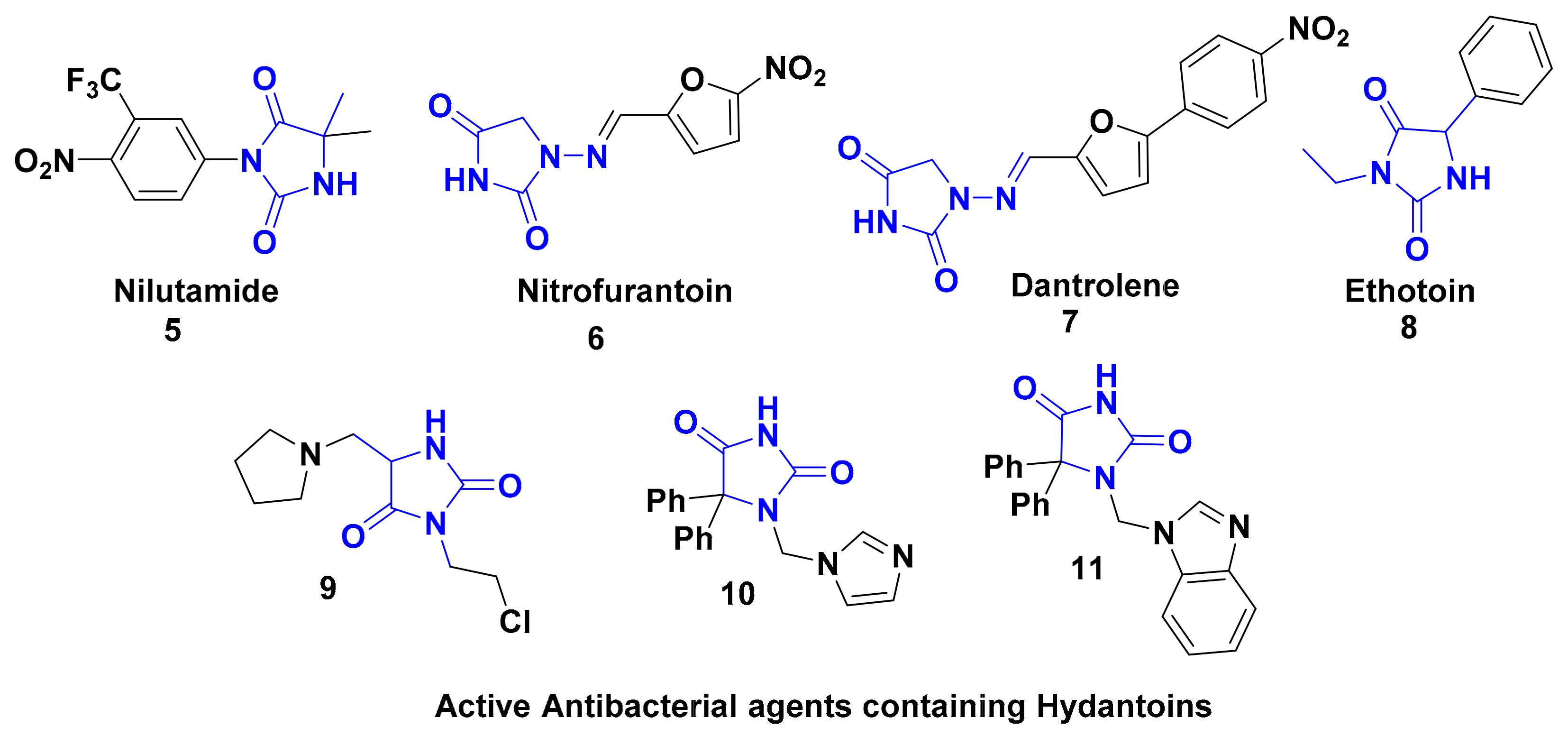
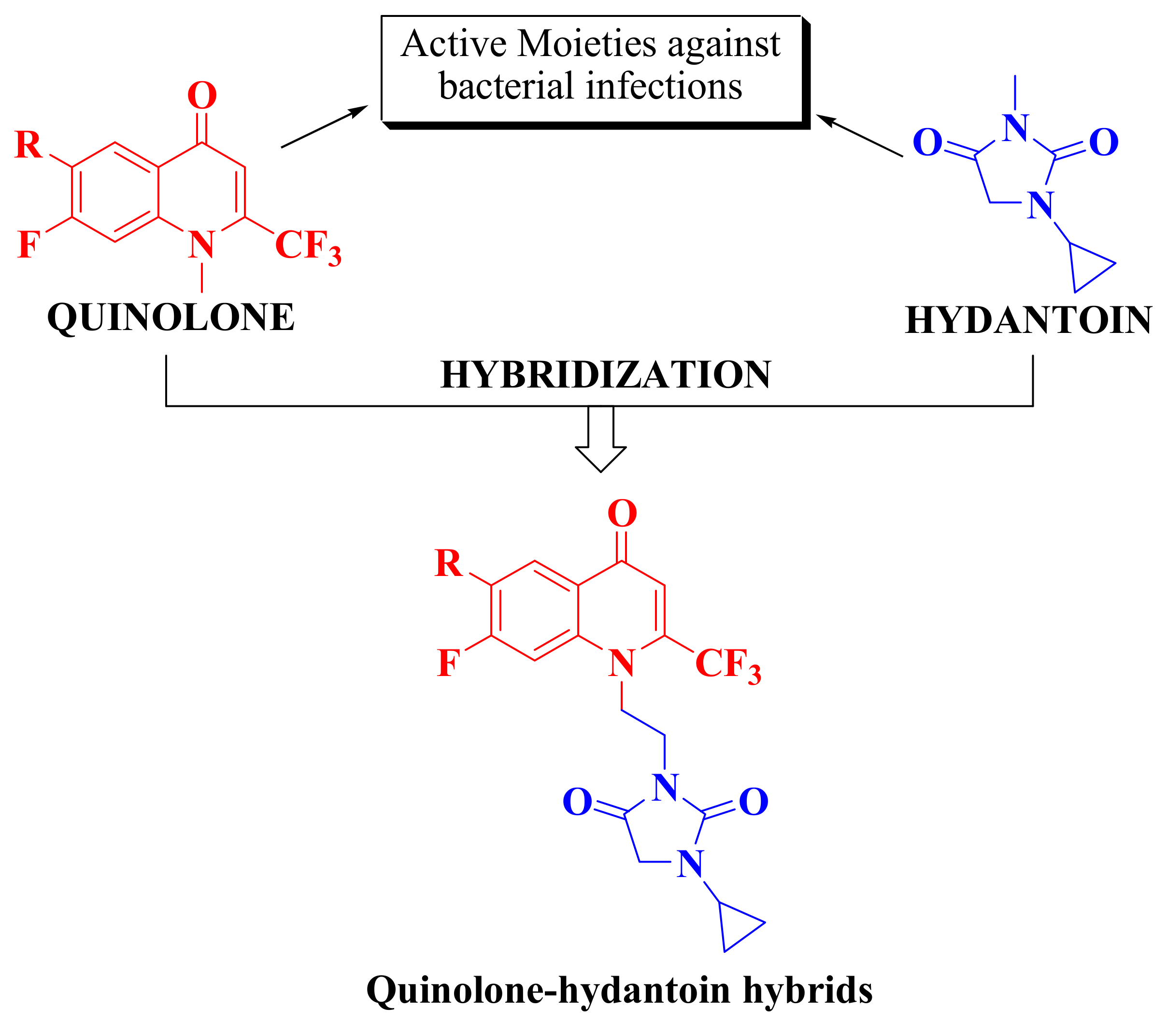
:1. Introduction
2. Results and Discussion
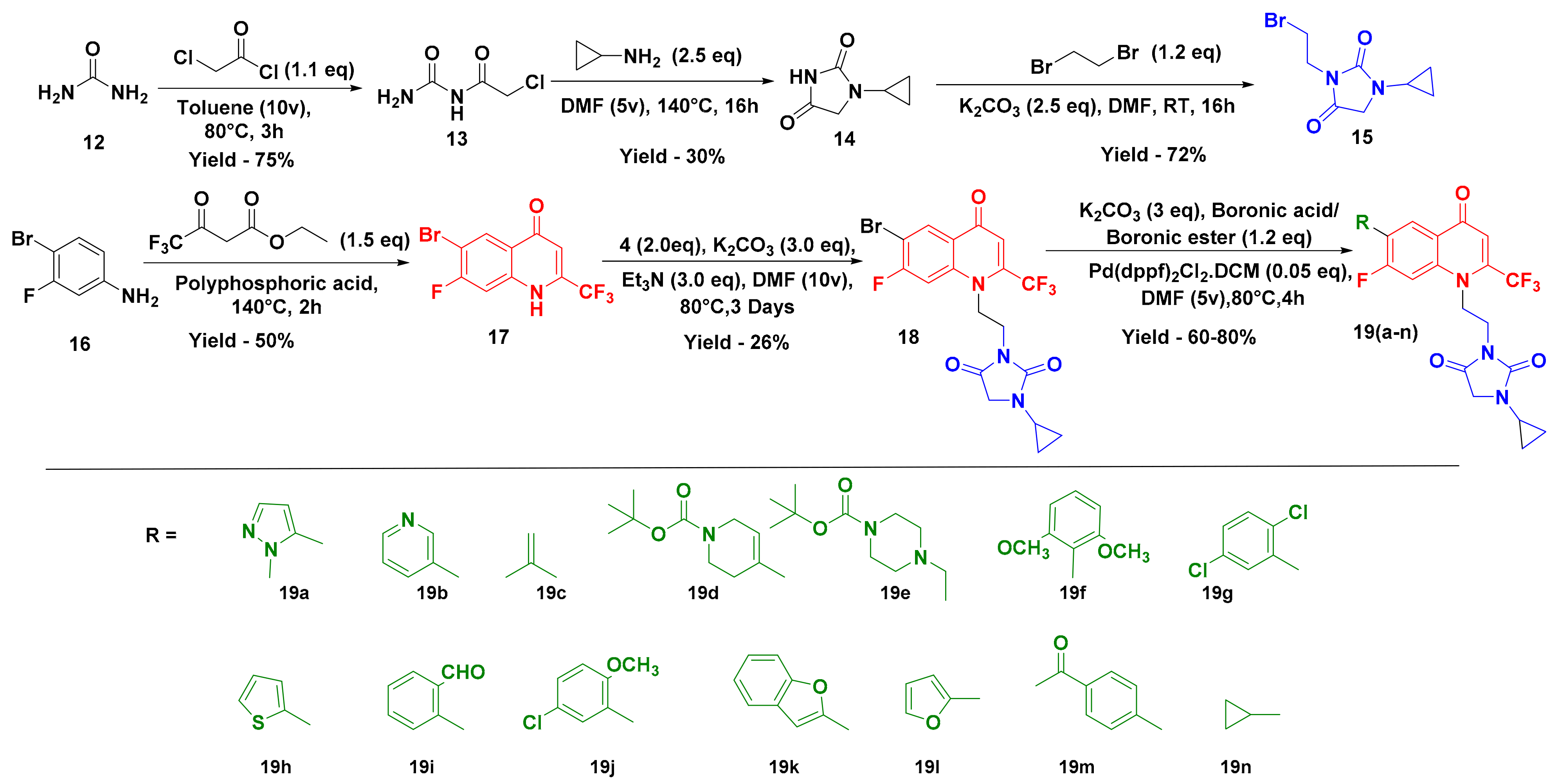
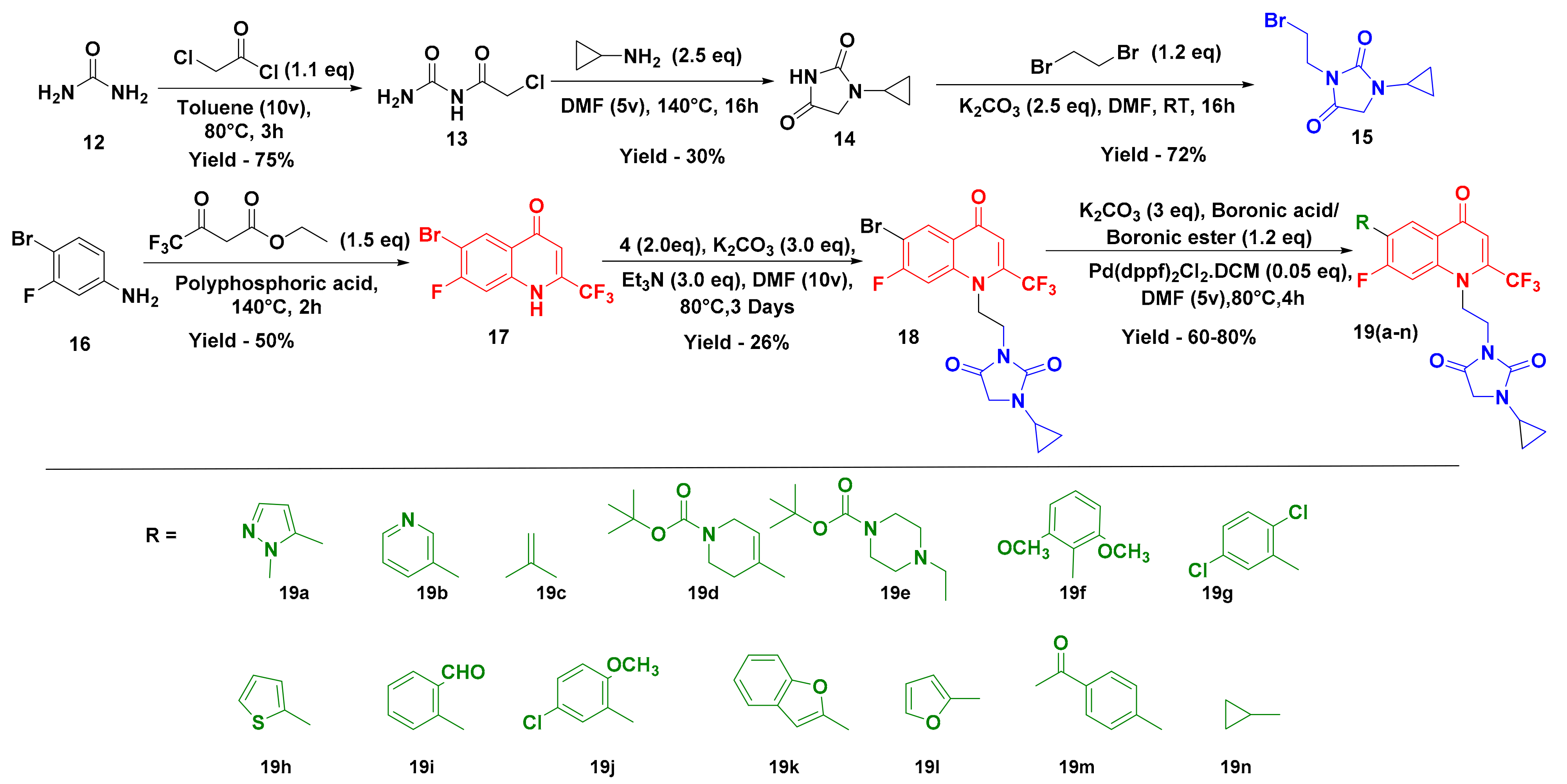
2.1. Chemistry
2.2. Biology
2.3. In Silico Phyicochemical Properties of Compound 19c
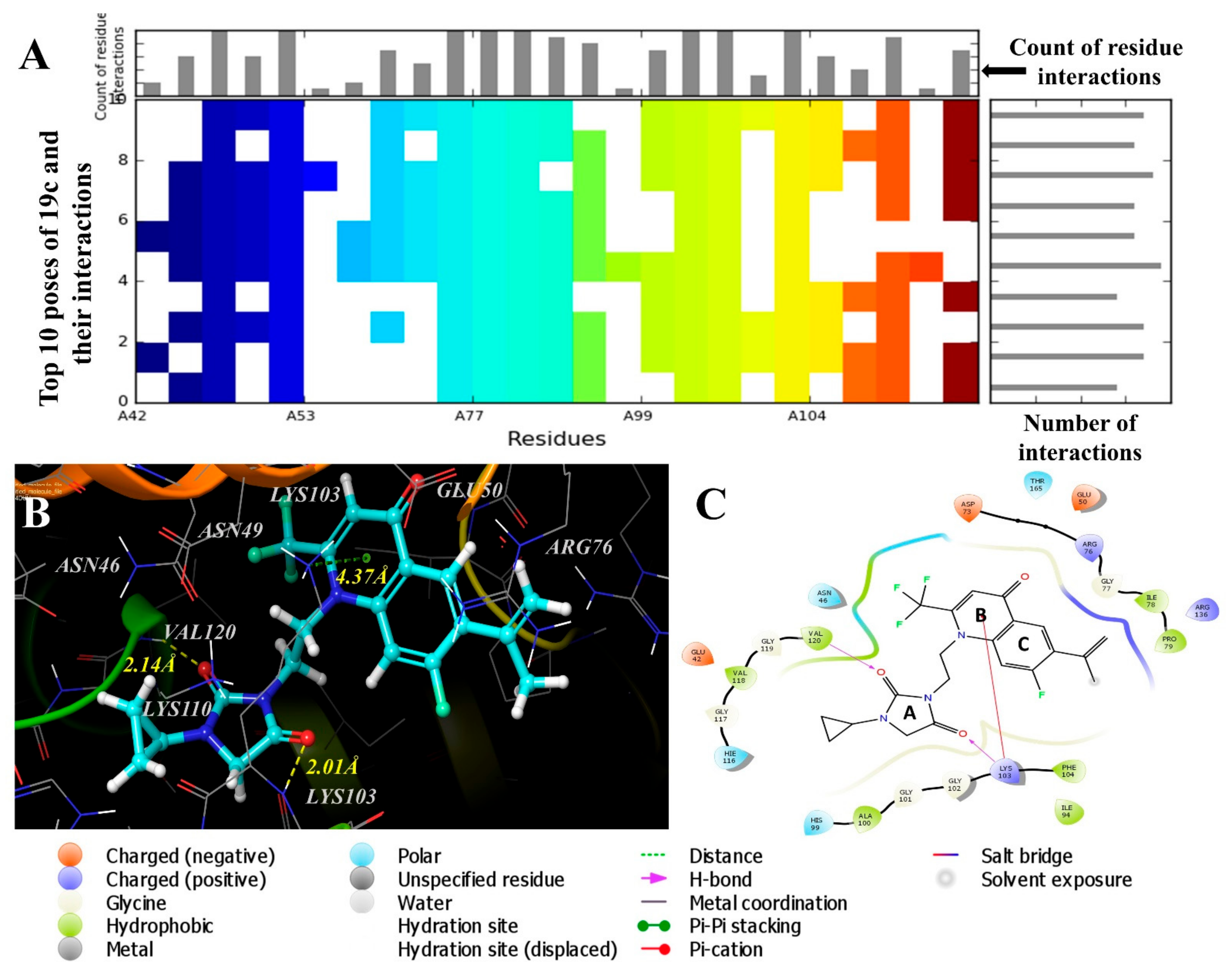
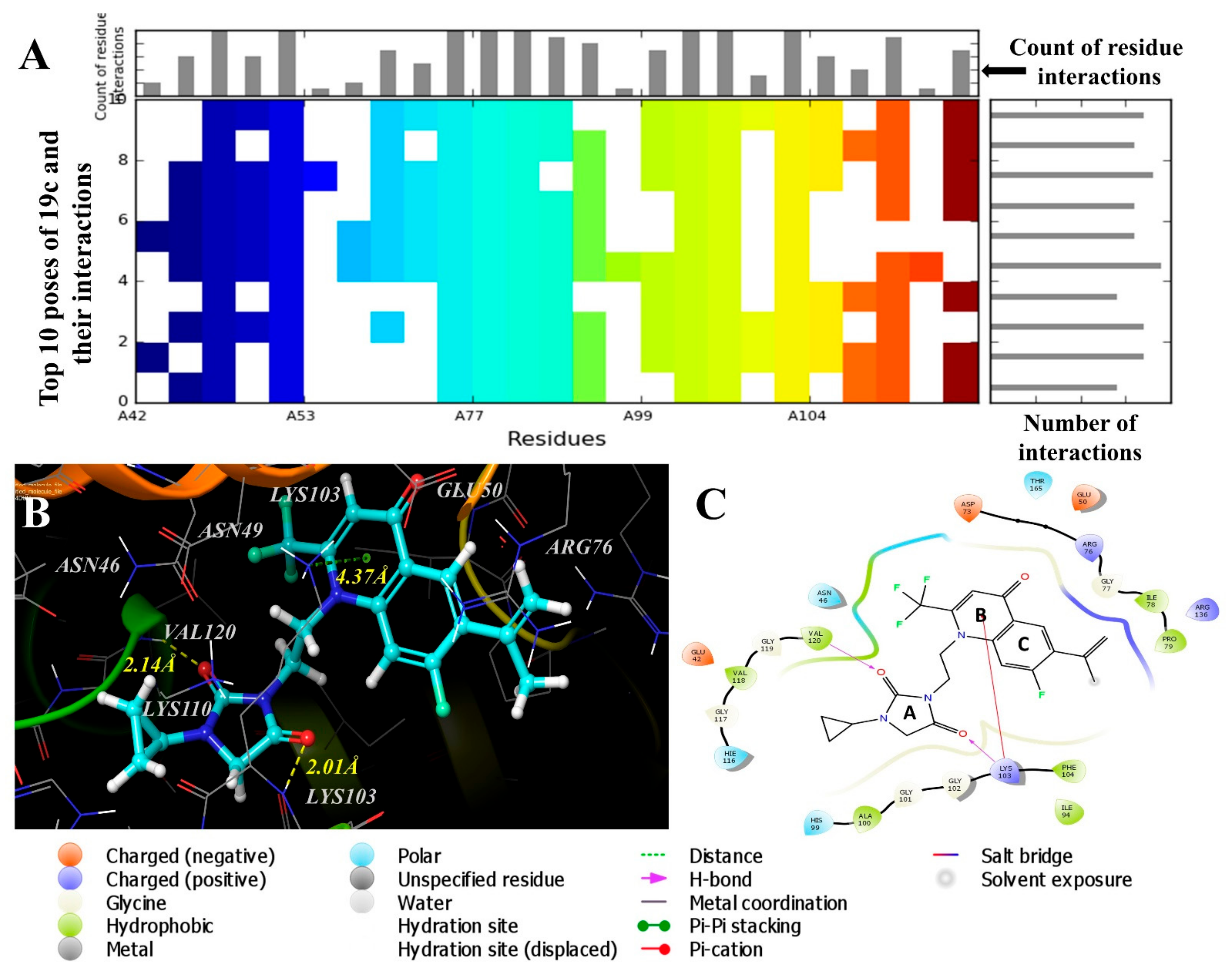
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. General Procedure
3.2. Antibacterial Activity
3.3. Agar Well Plate Method
3.4. Molecular Modeling Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chu, D.T.W.; Plattner, J.J.; Katz, L.J. New directions in Antibacterial Research. Med.Chem. 1996, 39, 3853–3874. [Google Scholar] [CrossRef]
- Beovic, B. The issue of an antimicrobial resistance in human medicine. Int. J. Food Microbiol. 2006, 112, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.; Hunter, P.A.J. Antibiotic resistance-Action to promote new technologies. Antimicrob. Chemother. 2006, 58, i3–i22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suree, N.; Jung, M.E.; Clubb, R.T. Recent advances toward new anti-infective agents that inhibit cell surface protein anchoring in staphylococcus aureus and other gram positive pathogens. Mini-Rev. Med.Chem. 2007, 7, 991–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitscher, L.A.; Pillai, S.P.; Gentry, E.J.; Shankel, D.M. Mutiple drug resistance. Med. Res. Rev. 1999, 19, 477–496. [Google Scholar] [CrossRef]
- Nikaido, H.; Zgurskaya, H.I. Antibiotic efflux mechanism. Curr. Opin. Infect. Dis. 1999, 12, 529–536. [Google Scholar] [CrossRef]
- WHO. 2021. Available online: https://www.who.int/news-room/detail/29-04-2019-new-report-calls-for-urgent-action-to-avert-antibacterial-resistance-crisis (accessed on 21 September 2021).
- Marepu, N.; Yeturu, S.; Pal, M. 1,2,3-Triazole fused with pyridine/pyrimidine as new template for antimicrobial agents: Regioselective synthesis and identification of potent N-heteroarenes. Bioorg. Med. Chem. Lett. 2018, 28, 3302–3306. [Google Scholar] [CrossRef]
- El-Gohary, N.-S.; Shaaban, M.I. Design, synthesis, antimicrobial, antiquorum-sensing and antitumor evaluation of new series of pyrazolopyridine derivatives. Eur. J. Med. Chem. 2018, 157, 729–742. [Google Scholar] [CrossRef]
- Gao, F.; Yang, H.; Lu, T.; Chen, Z.; Ma, L.; Xu, Z.; Schaffer, P.; Lu, G. Design, synthesis and anti-mycobacterial activity evaluation of benzofuran-isatin hybrids. Eur. J. Med. Chem. 2018, 159, 277–281. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Wang, X.; Wang, Y.; Tang, W.-J.; Shi, J.-B.; Liu, X.-H. Novel curcumin analogue hybrids: Synthesis and anticancer activity. Eur. J. Med. Chem 2018, 156, 493–509. [Google Scholar] [CrossRef]
- Maestro, A.; Martín-Encinas, E.; Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Vicario, J.; Palacios, F. Synthesis of novel antiproliferative hybrid bis-(3-indolyl)methane phosphonate derivatives. Eur. J. Med. Chem. 2018, 158, 874–883. [Google Scholar] [CrossRef]
- Chopra, R.; Chibale, K.; Singh, K. Pyrimidine-chloroquinoline hybrids: Synthesis and antiplasmodial activity. Eur. J. Med. Chem. 2018, 148, 39–53. [Google Scholar] [CrossRef]
- Mott, B.T.; Cheng, K.C.C.; Guha, R.; Kommer, V.P.; Williams, D.L.; Vermeire, J.J.; Cappello, M.; Maloney, D.J.; Rai, G.; Jadhav, A.; et al. A furoxan-amodiaquine hybrid as a potential therapeutic for three parasitic diseases. MedChemComm 2012, 3, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Saini, A.; Kumar, S.; Raj, R.; Chowdhary, S.; Gendrot, M.; Mosnier, J.; Fonta, I.; Pradines, B.; Kumar, V. Synthesis and antiplasmodial evaluation of 1H-1, 2, 3-triazole grafted 4-aminoquinoline-benzoxaborole hybrids and benzoxaborole analogues. Bioorg. Chem. 2021, 109, 104733. [Google Scholar] [CrossRef]
- King, D.E.; Malone, R.; Lilley, S.H. New classification and update on the quinolone antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar]
- Ryu, C.K.; Sun, Y.J.; Shim, J.Y.; You, H.J.; Choi, K.U.; Lee, H. Synthesis and antifungal activity of 6,7-bis-[S-(aryl)thio]-5,8-quinolinediones. Arch. Pharm. Res. 2002, 25, 795–800. [Google Scholar] [CrossRef]
- Musiol, R.; Jamilek, J.; Buchta, V.; Silva, L.; Niedbala, H.; Podeszwa, B.; Palka, A.; Maniecka, K.M.; Oleksyn, B.; Polanski, J. Antifungal properties of new series of quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 3592–3598. [Google Scholar] [CrossRef]
- Desai, U.; Mitragotri, S.; Thopate, T.; Pore, D.; Wadgaonkarb, P. A highly efficient synthesis of trisubstitutedquinolines using sodium hydrogensulfate on silica gel as a reusable catalyst. Arkivoc 2006, 15, 198–204. [Google Scholar]
- Nilsen, A.; LaCrue, A.N.; White, K.L.; Forquer, I.P.; Cross, R.M.; Marfurt, J.; Mather, M.W.; Delves, M.J.; Shackleford, D.M.; Saenz, F.E.; et al. Quinolone-3-diarylethers: A new class of antimalarial drug. Sci Trans. Med. 2013, 5, 177ra37. [Google Scholar] [CrossRef] [Green Version]
- Vu, A.T.; Cohn, S.T.; Manas, E.S.; Harris, H.A.; Mewshaw, R.E. ERβ ligands. Part 4: Synthesis and structure–activity relationships of a series of 2-phenylquinoline derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4520–4525. [Google Scholar] [CrossRef]
- Gogoi, S.; Shekarrao, K.L.; Duarah, A.; Bora, T.C.; Boruah, R.C. A microwave promoted solvent-free approach to steroidal quinolines and their in vitro evaluation for antimicrobial activities. Steriods 2012, 77, 1438–1445. [Google Scholar] [CrossRef]
- Ranu, B.C. Indium Metal and Its Halides in Organic Synthesis: Microreview. Eur. J. Org. Chem. 2000, 2000, 2347–2356. [Google Scholar] [CrossRef]
- Prajapati, S.M.; Patel, K.D.; Vekariya, R.H.; Panchal, S.N.; Patel, H.D. Recent advances in the synthesis of quinolines: A review. RSC Adv. 2014, 4, 24463–24467. [Google Scholar] [CrossRef]
- Amii, H.; KishiKawa, Y.; Uneyama, K. Rh(I)-Catalyzed Coupling Cyclization of N-Aryl Trifluoroacetimidoyl Chlorides with Alkynes: One-Pot Synthesis of Fluorinated Quinolines. Org. Lett. 2001, 3, 1109–1112. [Google Scholar] [CrossRef]
- Mahajan, A.; Chudawat, T.S. Review on the role of the metal catalyst for synthesis of pharmacologically important quinoline substrate. Mini Rev. Org. Chem. 2019, 16, 631–652. [Google Scholar] [CrossRef]
- McNaughton, B.R.; Miller, B.L. A mild and efficient one-step synthesis of quinolines. Org. Lett. 2003, 5, 4257–4259. [Google Scholar] [CrossRef]
- Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Recent advances in the synthesis of hydantoins: The state of the art of a valuable scaffold. Chem. Rev. 2017, 117, 13757–13809. [Google Scholar] [CrossRef]
- Lopez, L.I.L.; Loera, D.D.; Avalos, E.R.; Galindo, A.S. Green synthesis of hydantoins and its derivatives. Mini Rev. Org. Chem. 2020, 17, 176–184. [Google Scholar] [CrossRef]
- Fujisaki, F.; Toyofuku, K.; Egami, M.; Ishida, S.; Nakamoto, N.; Kashige, N.; Miake, F.; Sumoto, K. Antibacterial activity of some 5- dialkylaminomethylhydantoins and related derivatives. Chem. Pharm.Bull. 2013, 61, 1090–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehrani, K.H.M.E.; Hashemi, M.; Hassan, M.; Kobarfard, F.; Mohebbi, S. Synthesis and antibacterial activity of Schiff bases of 5-substituted isatins. Chin. Chem. Lett. 2016, 27, 221–225. [Google Scholar] [CrossRef]
- Bapna, M.; Parashar, B.; Sharma, V.K.; Chouhan, L.S. Microwave-Assisted synthesis of some novel and potent antibacterial and antifungal compounds with biological screening. Med. Chem. Res. 2012, 7, 1098–1106. [Google Scholar] [CrossRef]
- Alaa, A.B.; Hanin, A.B.; Safwat, A.A.; Reem, M.D.; Sameh, S.E. New bromoindole alkaloid isolated from the marine spongeHyrtios erectus. Heterocycles 2018, 96, 749–756. [Google Scholar]
- Nagai, T.; Nishioka, G.; Koyama, M.; Ando, A.; Miki, T.; Kumadaki, I. Reactions of trifluoromethyl ketones. IX. Investigation of the steric effect of a trifluoromethyl group based on the stereochemistry on the dehydration of trifluoromethyl homoallyl alcohols. J. Fluorine Chem. 1992, 57, 229–237. [Google Scholar] [CrossRef]
- Bravo, P.; Dillido, D.; Resnati, G. An efficient entry to perfluoroalkyl substituted azoles starting from β-perfluoroalkyl-β-dicarbonyl compounds. Tetrahedron 1994, 50, 8827–8836. [Google Scholar] [CrossRef]
- Gregory, T.P.; Darlene, C.D.; David, M.S. Structure-Activity Relationships for the Action of Dihydropyrazole Insecticides on Mouse Brain Sodium Channels Pestic. Biochem. Phys. 1998, 60, 177–185. [Google Scholar]
- Kucukguzel, S.G.; Rollas, S.; Erdeniz, H.; Kiranz, A.C.; Ekinci, M.; Vidin, A. Synthesis, characterization and pharmacological properties of some 4-arylhydrazono-2-pyrazoline-5-one derivatives obtained from heterocyclic amines. Eur. J. Med. Chem. 2000, 35, 761–771. [Google Scholar] [CrossRef]
- Marull, M.; Schlosser, M. Selective and efficient structural elaboration of 2-(trifluoromethyl)quinolinones. Eur. J. Org. Chem. 2003, 8, 1576–1588. [Google Scholar] [CrossRef]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. Fundam.Mol. Mech. Mutagen. 2007, 623, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Rizk, O.H.; Bekhit, M.G.; Hazzaa, A.A.B.; El-Khawass, E.M.; Abdelwahab, I.A. Synthesis, antibacterial evaluation and DNA gyrase inhibition profile of some newquinoline hybrids. Arch. Pharm. 2019, 352, 1900086. [Google Scholar] [CrossRef]
- Omar, A.M.; Alswah, M.; Ahmed, H.E.A.; Bayoumi, A.H.; El-Gamal, K.M.; El-Morsy, A.; Ghiaty, A.; Afifi, T.H.; Sherbiny, F.F.; Mohammed, A.S.; et al. Antimicrobial screening and pharmacokinetic profiling of novel phenyl-[1,2,4]triazolo[4,3-a] quinoxaline analogues targeting DHFR and E. coli DNA gyrase B. Bioorg. Chem. 2020, 96, 103656. [Google Scholar] [CrossRef]
- Hooper, D.C.; Jacoby, G.A. Topoisomerase inhibitors: Fluoroquinolone mechanisms of action and resistance. Perspect. Med. 2016, 6, a025320. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Valgas, C.; Souza, S.M.D.; Smânia, E.F.; Smânia, A. Screening methods to determine antibacterial activity of natural products. Braz. J. Microbiol. 2007, 38, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Arya, A.; Gupta, K.; Chundawat, T.S.; Vaya, D. Biogenic synthesis of copper and silver nanoparticles using green alga Botryococcus braunii and its antimicrbial activity. Bioinorg. Chem. Appl. 2018, 2018, 7879403. [Google Scholar] [CrossRef] [Green Version]
- DeAlba-Montero, I.; Guajardo-Pacheco, J.; Morales-Sánchez, E.; Araujo-Martínez, R.; Loredo-Becerra, G.M.; Martínez-Castañón, G.A.; Compeán Jasso, M.E. Antimicrobial properties of copper nanoparticles and amino acid chelated copper nanoparticles produced by using a soya extract. Bioinorg. Chem. Appl. 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665. [Google Scholar] [CrossRef] [Green Version]
- LigPrep, Version 2.3; Schrodinger, LLC.: New York, NY, USA, 2009.
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-Based Discovery of Substituted 4,5′-Bithiazoles as Novel DNA Gyrase Inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | S. aureus | K. pneumoniae | P. aeruginosa | E. coli |
|---|---|---|---|---|
| 19a | 18 ± 1.23 | 16 ± 1.42 | 12 ± 1.22 | 12 ± 0.09 |
| 19b | 10 ± 1.44 | 12 ± 0.72 | 12 ± 0.87 | 12 ± 0.47 |
| 19c | 18 ± 0.89 | 20 ± 0.88 | 14 ± 0.76 | 16 ± 1.33 |
| 19d | 15 ± 0.87 | 15 ± 0.77 | 13 ± 0.23 | 12 ± 1.45 |
| 19e | 12 ± 0.76 | 13 ± 0.68 | 12 ± 0.55 | 10 ± 1.77 |
| 19f | 20 ± 0.88 | 18 ± 1.76 | 16 ± 0.16 | 14 ± 1.67 |
| 19g | 20 ± 0.59 | 17 ± 1.23 | 16 ± 0.34 | 14 ± 0.67 |
| 19h | 16 ± 1.45 | 16 ± 1.11 | 12 ± 1.55 | 12 ± 0.35 |
| 19i | 17 ± 1.23 | 18 ± 1.35 | 16 ± 1.87 | 14 ± 0.13 |
| 19j | 13 ± 0.93 | 14 ± 1.66 | 15 ± 1.15 | 13 ± 0.77 |
| 19k | 21 ± 0.86 | 20 ± 1.64 | 16 ± 1.09 | 12 ± 0.89 |
| 19l | 18 ± 1.56 | 16 ± 0.99 | 14 ± 0.53 | 14 ± 0.45 |
| 19m | 12 ± 1.76 | 22 ± 1.43 | 18 ± 0.35 | 16 ± 1.45 |
| 19n | 18 ± 1.87 | 20 ± 1.65 | 16 ± 0.88 | 14 ± 1.23 |
| Chloramphenicol | 22 ± 0.33 | 25 ± 0.75 | 20 ± 0.16 | 21 ± 0.66 |
| Ampicillin | 24 ± 0.16 | 19 ± 1.21 | - | 18 ± 1.44 |
| Compound | S. aureus | K. pneumoniae | P. aeruginosa | E. coli |
|---|---|---|---|---|
| 19a | 200 | 125 | 100 | 100 |
| 19b | 200 | 200 | 100 | 100 |
| 19c | 50 | 100 | 50 | 100 |
| 19d | 200 | 100 | 100 | 250 |
| 19e | 250 | 125 | 100 | 100 |
| 19f | 125 | 100 | 250 | 200 |
| 19g | 500 | 250 | 100 | 100 |
| 19h | 100 | 100 | 250 | 100 |
| 19i | 50 | 250 | 62.5 | 50 |
| 19j | 125 | 50 | 250 | 200 |
| 19k | 100 | 200 | 50 | 125 |
| 19l | 250 | 200 | 100 | 125 |
| 19m | 125 | 100 | 250 | 200 |
| 19n | 250 | 100 | 100 | 250 |
| Chloramphenicol | 50 | 50 | 50 | 50 |
| Ampicillin | 25 | 100 | - | 100 |
| Compound | M.wt | No. of H-Bond Accepters | No. of H-Bond Donors | Log P | Molar Refractivity | No. of Lipinski Violations |
|---|---|---|---|---|---|---|
| 19c | 437 | 7 | 0 | 4.0 | 112.31 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahajan, A.; Singh, H.; Singh, A.; Agrawal, D.K.; Arora, A.; Chundawat, T.S. Trifluoromethylated Quinolone-Hydantoin Hybrids: Synthesis and Antibacterial Evaluation. Sci 2022, 4, 30. https://doi.org/10.3390/sci4030030
Mahajan A, Singh H, Singh A, Agrawal DK, Arora A, Chundawat TS. Trifluoromethylated Quinolone-Hydantoin Hybrids: Synthesis and Antibacterial Evaluation. Sci. 2022; 4(3):30. https://doi.org/10.3390/sci4030030
Chicago/Turabian StyleMahajan, Akhil, Harbinder Singh, Amandeep Singh, Devendra K. Agrawal, Amandeep Arora, and Tejpal Singh Chundawat. 2022. "Trifluoromethylated Quinolone-Hydantoin Hybrids: Synthesis and Antibacterial Evaluation" Sci 4, no. 3: 30. https://doi.org/10.3390/sci4030030
APA StyleMahajan, A., Singh, H., Singh, A., Agrawal, D. K., Arora, A., & Chundawat, T. S. (2022). Trifluoromethylated Quinolone-Hydantoin Hybrids: Synthesis and Antibacterial Evaluation. Sci, 4(3), 30. https://doi.org/10.3390/sci4030030