Abstract
A series of new trifluoromethyl-substituted quinolones and hydantoin hybrids has been synthesized and evaluated against Gram-positive bacterium (Staphylococcus aureus MTCC 96) and Gram-negative bacteria (Pseudomonas aeruginosa MTCC 441, Klebsiella pneumonia MTCC 109, and Escherichia coli MTCC 442). Compound 19c, having the 6-propene group on the quinolone ring, showed similar activity to a standard drug (chloramphenicol) by exhibiting MIC values of 50 µg/mL against S. aureus and P. aeruginosa. Physicochemical properties of compound 19c were also determined, which were in line with Lipinski’s rule of five, suggesting the suitability of compound 19c in biological systems. Various types of binding interactions of 19c within the active site of DNA gyrase of S. aureus were also streamlined by molecular docking studies, suggesting its capability to block the catalytic process of the DNA gyrase, which could be the possible reason for its antibacterial potential.
1. Introduction
Microbial resistance is becoming a severe threat to human society [1,2,3]. According to the World Health Organization (WHO) report, at least 50,000 people die daily due to drug-resistant diseases globally, including in developed and developing countries [4]. The most familiar terminology used to describe these drug-resistant pathogens is “superbugs”. Several efforts in developing new antibacterial agents are insufficient to meet the expectations due to increasing microbial resistance and not having a good toxicity profile of antibacterial agents. As a result, there is a continuous need to develop new antibacterial agents [5]. Several research strategies have been utilized to tackle the antibacterial drug resistance problem. It is often observed that drug resistance is more problematic in solo-targeting agents, resulting from the failure of an expected drug candidate [6]. However, a drug that targets several sites instead of a single site will result in greater potency, minimum resistance, and less toxicity [7,8,9,10]. Molecular hybridization is one of the most prominent strategies in developing novel drug entities and is the most commonly used to tackle such problems these days.
Molecular hybridization combines two active pharmacophores and results in a new single entity with improved efficacy compared to the parent drug. In addition, drug hybridization has proven advantageous in terms of increased drug efficacy, reduced cytotoxicity, and most importantly, decreased resistance compared to the parent drug [11]. For example, several promising antiplasmodial hybrids were synthesized with quinolone as the core moiety, artemisinin-4-aminoquinoline, clotrimazole-quinoline, 4-aminoquinoline-phthalimide, 4-aminoquinoline-naphthalimide, 4-aminoquinoline-prymidine, 4-aminoquinoline-purine, etc. [12].
Quinolone and its fluorine derivatives are nitrogen-rich heterocyclic compounds. They are broad-spectrum antibiotics and possess high oral bioavailability with excellent antibacterial potential. However, their clinical use is minimal because of their ability to produce serious side effects. So far, four generations of quinolines are available to humans, and few analogs have been commercialized, including ciprofloxacin (1), levofloxacin (2), norfloxacin (3), gatifloxacin (4), etc. (Figure 1) [13]. Other than antibacterial activity, quinolone analogs are known to exhibit other therapeutic activities, such as antimalarial [14,15,16,17], antifungal [18,19], antiplatelet [20], anticancer [21,22], and antitubercular [23,24].
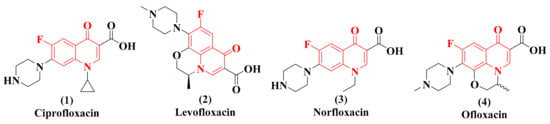
Figure 1.
Commercialized examples of biologically active quinolone derivatives.
Hydantoin, a heterocyclic organic compound, is well-known for its biological properties. Many commercial products, such as Phenytoin, Ethotoin (8), Fosphenytoin, Nitrofurantoin (6), Dantrolene (7), Nilutamide (5), etc. [25,26] (Figure 2), are known to exhibit versatile therapeutic properties. In the recent past, a number of significant research reports have been published justifying the antibacterial potential of hydantoin derivatives. For example, Fujisaki et al. reported the antibacterial activity of 5-dialkyl aminomethyl hydantoins and its analogs [27]. Haj Mohammad et al. reported the synthesis and antibacterial activity of Schiff bases of 5-substituted isatins (9) [28]. Bapna et al. reported that 3,5,5-triphenylimidazolidine-2,4-dione and 5,5-diphenyl imidazolidine-2,4-dione showed good antibacterial activity (10, 11) [29]. Alaa et al. reported that 5-((6-Bromo-1H-indol-3-yl)methyl)-3-methyl imidazolidine-2,4-dione isolated from marine sponge Hyrtios erectus showed moderate antibacterial activity against S. aureus and E. coli [30] (Figure 2).
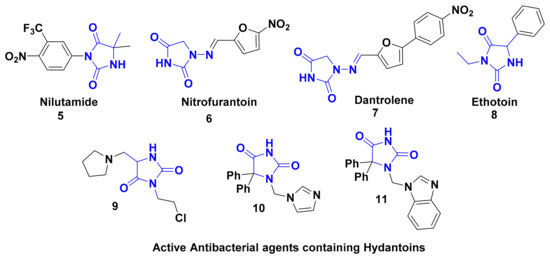
Figure 2.
Commercialized and other representative examples of biologically active hydantoin derivatives.
Halogens, specifically fluorine substituents, are the focus of much interest in drug design. Fluorine increases lipophilicity, changes the physicochemical properties, enhances stability, and improves antibacterial activity, resulting in therapeutic efficiency [31]. Having substitution with a trifluoromethyl group could also increase therapeutic efficiency. An increase in size and volume results higher in hydrophobicity than in the case when fluorine is present. In addition, several biologically active molecules with trifluoromethyl groups are herbicides [32], antipyretics [33], and platelet aggregation inhibitors [34]. Fluorine-substituted quinolone antibiotic drugs available in the market are Ciprofloxacin, Levofloxacin, Norfloxacin, and Gatifloxacin. Due to excellent profile in the area of antibacterial drugs, trifluoromethyl group has been selected to include into a target hybrid molecule.
Considering the importance of global health concerns of drug-based resistance as a major hurdle of available antibacterial drugs, there is an urgent need to develop novel potent antibacterial agents. In this present study, we targeted the synthesis and characterization of trifluoromethylated quinolones tethered with hydantoin hybrids (Figure 3). We evaluated newly synthesized hybrids against several bacterial strains by checking their zones of inhibition and calculated their minimum inhibitory concentration values. Furthermore, molecular docking studies were used to explore several binding interactions of potent compounds with DNA gyrase.
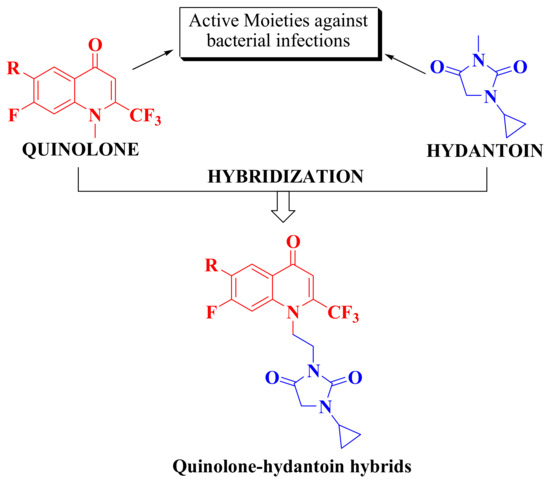
Figure 3.
Design strategy of trifluoromethylated quinolone-tethered hydantoin hybrids.
2. Results and Discussion
2.1. Chemistry
Our studies commenced by synthesizing the targeted compounds in six steps. Initially, commercially available urea (12) was treated with chloroacetylchloride in the presence of toluene to obtain carbamoyl-2-chloroacetamide (13), which was further refluxed with cyclopropylamine in DMF for 16 h to obtain 1-cyclopropylimidazolidine-2,4-dione (14). The treatment of compound 14 with 1,2-dibromoethane in the presence of K2CO3 using DMF as a solvent for 16 h at room temperature resulted in 3-(2-bromomethyl)-1-cyclopropylimidazolidine-2,4-dione (15).
Simultaneously, 6-bromo-7-fluoro-2-(trifluoromethyl)-quinolin-4(1H)-one (17) was prepared by intermolecular condensation of 4-bromo-3-fluoroaniline (16) with ethyl 4,4,4-trifluoro-3-oxobutanoate in polyphosphoric acid at 130 °C for 2 h following Marull’s procedure [35]. Then, 3-(2-(6-Bromo-7-fluoro-4-oxo-2-(trifluoromethyl)quinoline-1(4H)-yl)ethyl)-1-cyclopropyl imidazolidine-2,4-dione (18) was obtained by treating 6-bromo-7-fluoro-2-(trifluoromethyl)quinoline-4(1H)-one (17) with 3-(2-bromomethyl)-1-cyclopropyl imidazolidine-2,4-dione (15) in the presence of K2CO3 and triethylamine as a base and DMF as a solvent at 80 °C for 48 h. Suzuki coupling using 3-(2-(6-bromo-7-fluoro-4-oxo-2-(trifluoromethyl) quinoline-1(4H)-yl)ethyl)-1-cyclopropyl imidazolidine-2,4-dione (18), boronic ester/boronic acid, Pd(dppf)Cl2.DCM, and K2CO3 in DMF at 80 °C was performed to yield compound 19 (Scheme 1). All the synthetics were structurally characterized by using spectral and analytical evidence. For example, compound 19c exhibited a molecular ion peak at 437.9 in its high-resolution mass spectrum (HRMS). Its 1H NMR spectrum showed the presence of a multiplet at δ 4.46–4.44 (2H) and a singlet at δ 3.91 (2H) due to the presence of an alkyl linker. The presence of two singlets at δ 2.25 (3H) and δ 0.67 (4H) corresponds to the methyl group and cyclopropane ring, which further confirmed the structure of compound 19c. The presence of characteristic peaks at δ 5.1 and 22.7 ppm corresponding to the cyclopropane ring and an α, β-unsaturated carbonyl carbon at δ 170.2 in the 13C NMR spectrum further corroborated the structure of the relevant compound.
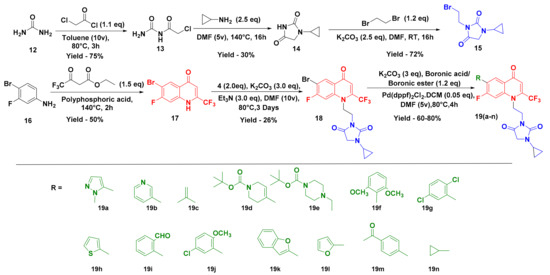
Scheme 1.
Synthesis of trifluoromethylated quinolones and hydantoin hybrids.
2.2. Biology
All the synthesized compounds (19) were evaluated for their antibacterial activity against one Gram-positive bacterium (Staphylococcus aureus MTCC 96) and three Gram-negative bacterial strains (Pseudomonas aeruginosa MTCC 441, Klebsiella pneumonia MTCC 109, and Escherichia coli MTCC 442). Ampicillin (active against Gram-positive bacteria) was used to compare the results of compounds against the Gram-positive bacteria and chloramphenicol (broad-spectrum antibiotic) was used to compare the results against both Gram-positive as well as Gram-negative bacterial strains.
The antibacterial potential of these compounds was monitored by determining the zone of inhibition (mm) against different bacterial strains, and the values are summarized in Table 1. Almost all the synthesized hybrids showed moderate to good activity against all the tested bacterial strains. Careful examination of Table 1 showed that four compounds, 19c, 19f, 19g, and 19k, exhibited comparable zones of inhibition to that of chloramphenicol, which is a broad-spectrum antibiotic. However, the zone of inhibition of these compounds was found smaller than that of the standard drug ampicillin that specifically targets the Gram-positive bacterium. Amongst the Gram-negative bacterial strains, almost all the compounds were found less active than standard drugs, except the K. pneumoniae bacterial strain, which was found to be the most sensitive one against these hybrid molecules. Four compounds, 19c, 19k, 19m, and 19n, showed comparable antibacterial potential to that of chloramphenicol against K. pneumoniae. Minimum inhibitory concentration (MIC) values were calculated for all the synthesized hybrids to further compare these compounds with their respective standard drugs (Table 2). Compound 19c, having a 6-propene group on the quinolone ring, displayed equal potency (MIC 50 µg/mL) to the standard drug chloramphenicol (MIC 50 µg/mL) against S. aureus, however it was found to be two-fold less active against S. aureus compared to the standard drug ampicillin, which is a specific antibiotic against Gram-positive bacteria. These biological results revealed an interesting structure–activity relationship (SAR) which concluded that the size of the substitution at the sixth carbon atom (R) of the quinolone moiety considerably influenced the antibacterial potential. Specifically, for the Gram-positive bacterial strain (S. aureus), the hybrid compounds substituted with small groups showed better activity than the compounds substituted with bulky groups. It is justified by the zone of inhibition values of hybrid compounds with R group as smaller groups, for example compounds 19a, 19c, 19k, 19l, and 19n have smaller groups which could have less steric hindrance as compared to that of the compounds substituted with bulkier groups, such as 19d, 19e, and 19m, and are the least active compounds against S. aureus. Hybrid compounds with bulkier groups at position R can experience steric hindrance that could make them fit less within the active site of DNA gyrase, which could be the reason for their lower activity than that of compounds with smaller groups at R position (later justified through molecular docking studies with DNA gyrase). An almost similar trend for SAR was observed with the zone of inhibition values of compounds against the Gram-negative bacterial strain K. pneumoniae. Although most of the compounds exhibited antibacterial activity against different strains, compound 19c with a propene group at the sixth position of quinolone (Table 2) seems to be a promising compound, having good inhibitory potential against both Gram-positive and Gram-negative bacterial strains. The broad-spectrum nature of this compound could have made it slightly more active than the standard drug ampicillin, but it had comparable potential to that of chloramphenicol.

Table 1.
Zone of inhibition (mm) of synthesized compounds, 19a–n, against Gram-positive and Gram-negative bacteria.
Table 2.
Minimum inhibitory concentration (µg/mL) of hybrids 19a–n against Gram-positive and Gram-negative bacteria.
2.3. In Silico Phyicochemical Properties of Compound 19c
The physicochemical properties of 19c were obtained using the web-based SwissADME (http://swissadme.ch/index.php (accessed on 25 February 2022)) to assess their compliance with the Lipinski rule of five criteria (Table 3). The analysis of data revealed that: (a) the molecular weight of 19c is 437, which is within the accepted limit of 500, (b) a log P lower than 5.0 was found, indicating that the compound was not very lipophilic, and (c) molar refractivity of 112.3 was found, which is within the accepted limits of 40–130. Compound 19c also complies with the hydrogen bond properties. The results of this analysis suggest that 19c show compliance with the Lipinski rule, which is known as the filters for the drug’s likeness to increase the efficiency of drug discovery, and thus represents a pharmacologically active framework that could be considered for progressing further potential hits.
Table 3.
In silico physicochemical properties of compound 19c.
2.4. Molecular Modeling Studies
DNA gyrase enzyme is a topoisomerase II, which is crucial for DNA transcription and replication processes in eukaryotic cells [36]. The inhibition of DNA gyrase has been considered as a striking goal for the development of antibacterial agents against various bacterial pathogens [37,38]. Quinolone antibiotics are an existing class of agents that have been clinically used to inhibit bacterial DNA synthesis [39,40]. Fluoroquinolones and their derivatives work as DNA gyrase poisons as they could inhibit bacterial nucleic acid synthesis, thereby leading to cell death [39]. It was expected that the synthesized quinolone-hydantoin hybrids could also inhibit the DNA gyrase, and therefore, molecular docking studies were performed to investigate the interactions between the most active compound 19c and DNA gyrase. Induced fit docking (IFD) is the validated protocol that accurately predicts ligand binding modes and structural changes in the protein or receptor on ligand binding. It is an advantageous tool of Schrodinger Suite over the rigid docking. Therefore, instead of rigid docking, we performed IFD of the compound 19c in the binding pocket of DNA gyrase (PDB entry: 4DUH; Resolution 1.50Å) and generated twenty poses with DNA gyrase with different dock scores and binding interactions.
We selected the top ten poses from the IFD results and generated an Interaction Fingerprint (Figure 4A) to analyze the number of interactions and number of interacting residues with each generated pose of 19c. Careful examination of Figure 4A concluded that pose 2 of 19c has a higher dock score (−10.091) with a consistent and high number of interactions with various amino acid residues in the binding site of DNA gyrase. Therefore, pose 2 was selected to further explain the binding of compound 19c with DNA gyrase.
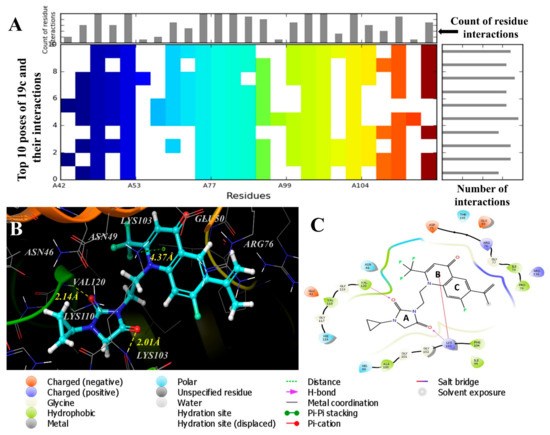
Figure 4.
(A) Top ranked poses generated through induced fit docking of compound 19c with DNA gyrase (PDB: 4DUH). (B) Top ranked pose selected to explain the binding of compound 19c with DNA gyrase in 3D representation. (C) 2D representation of compound 19c with DNA gyrase.
It was found that 19c fits well at the binding site and is stabilized by H-bonds and polar interactions with various amino acid residues (Figure 4B,C). Interestingly, Ring A (hydantoin) is well-occupied in a cavity formed by hydrophobic residues Val120, Val118, and Ala100, and polar residues His99, His116, and Asn46, in which various glycine amino acids are also involved to stabilize the Ring A. The cyclopropyl group attached to the N-atom is placed deeply into this cavity and helps to stabilize the Ring A (hydantoin moiety). Both the carbonyl O-atoms of Ring A are involved in H-bond interactions with the amino acid residues of Val120 and Lys103, with the distances of 2.14Å and 2.01Å, respectively (Figure 4B, C). These two hydrogen bonds showed their significance in the tight binding of compound 19c with DNA gyrase. Similarly, Ring B and Ring C (quinolone rings) are well-occupied by various positively and negatively charged amino acid residues, such as Asp73 and Glu50 (negatively charged), and Arg76 and Arg136 (positively charged) (Figure 4C). Ring B of quinolone is involved in the π-cation interaction with Lys103 amino acid residue with the distance of 4.37Å (Figure 4B, C). The bulky alkyl group present on Ring C also helps to stabilize the molecule in the binding pocket of DNA gyrase through hydrophobic interactions with Ile78 and Ile79 (Figure 4C). These binding interactions conclude that DNA gyrase is the probable target of these hybrid molecules, the inhibition of which is responsible for their antibacterial action.
3. Materials and Methods
3.1. General Procedure
All the required chemicals were purchased from Spectrochem and Aldrich Chemical Company. Pre-coated aluminum sheets (silica gel 60 F254, Merck, Kenilworth, NJ, USA) were used for thin-layer chromatography (TLC) and spots were visualized under UV light. 1H and 13C NMR spectra were recorded on an Agilent Technologies 400 MHz VNMRS spectrometer (400/100 MHz) using DMSO as the internal standard. Chemical shifts (δ) are reported in parts per million (ppm). The coupling constants (J) are reported in Hz. Mass spectrometric data were obtained with a Finnigan LCQ-Classic (ESI-MS; Thermo Separation Products, San José, CA, USA) or an expression S compact mass spectrometer (APCI-MS; Advion Inc., Ithaca, NY, USA).
Synthesis and characterization of N-carbamoyl-2-chloroacetamide (13)
To a solution of urea (50 g, 0.832 moles, 1.0 equiv) in toluene (500 mL, 10 v), chloroacetyl chloride (79.6 mL, 0.999 moles, 1.2 equiv) was added at room temperature under N2 atmosphere. The reaction mass was stirred at ambient temperature for 10 min and heated to 80 °C for 3 h. Completion of the reaction was confirmed by 1H NMR analysis. After completion of the reaction, the RM (reaction mixture) was poured over the Buchner funnel under reduced pressure to obtain white solids washed with toluene (10 v). The obtained solid material was further stirred in ice-cold water (5 v) for 10 min and then filtered to obtain the desired material as a white solid of 85.26 g, with percent yield 75%, which was further dried in an oven. M. Pt (188 °C); 1H NMR (400 MHz, DMSO-d6) δ10.40 (s, NH, 1H), 7.48–7.35 (m, NH2, 2H), 4.26 (s, CH2, 2H); 13C NMR (400 MHz, DMSO) δ168.09, 153.42, 43.23; MS (m/z) calculated: 136.54, MS (m/z) observed: no ionization observed.
Synthesis of 1-cyclopropylimidazolidine-2,4-dione (14)
To a solution of N-carbamoyl-2-chloroacetamide (85 g, 0.622 moles, 1.0 equiv) in DMF (425 mL, 5 v), cyclopropylamine (107.83 mL, 1.55 moles, 2.5 equiv) was added dropwise under N2 atmosphere. Exothermicity was observed during the addition of cyclopropylamine. The reaction mass was stirred at 40 °C for 6 h and then at 140 °C for 16 h. Completion of the reaction was confirmed by TLC and mass analysis. After completion of the reaction, the RM was brought to 60 °C and then concentrated under reduced pressure to obtain crude material, and then further purified using column chromatography (60–120 mesh silica) to obtain the desired material in 20% acetone/DCM. The desired material was further stirred in diethyl ether (10 v) to obtain a white solid. The yield obtained was 26.17 g, percent yield 30%, white solid; M.pt (135 °C); 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, NH, 1H), 3.86 (s, CH2, 2H), 2.57–2.54 (m, CH (cyclopropyl), 1H), 0.69–0.65 (m, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 171.82, 157.31, 51.21, 24.30, 5.26,; MS (m/z) calculated: 140.14, MS (m/z) observed: [M+H]+: 141.02.
Synthesis of 3-(2-bromoethyl)-1-cyclopropyl imidazolidine-2,4-dione (15)
To a solution of 1-cyclopropylimidazolidine-2,4-dione (26 g, 0.185 moles, 1.0 equiv) in DMF (260 mL, 10 v), K2CO3 (64.10 g, 0.463 moles, 2.5 equiv) was added followed by addition of 1, 2-dibromoethane (19.27, 0.222 moles, 1.2 equiv) under N2 atmosphere. The reaction mass was stirred at rt for 16 h. TLC confirmed the completion of the reaction. After completion of the reaction, ethyl acetate/water workup was performed. The organic layer was washed with 15% brine (10 v × 3) to get rid of DMF. Finally, the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to obtain the desired material as oil, which was used as such in the next step without further purification, with an obtained yield of 33 g, percent yield 72%, liquid; 1H NMR (400 MHz,DMSO-d6) δ 3.96 (s, CH2, 2H), 3.73 (t, J = 8.00 Hz, CH2, 2H), 3.59 (t, J = 8.00 Hz, CH2, 2H), 2.66–2.60 (m, CH (cyclopropyl),1H), 0.74–0.65 (m, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 169.86, 155.99, 49.87, 32.010, 29.22, 24.52, 5.10 MS (m/z) calculated: 247.09, MS (m/z) observed: [M+H]+: 247.0.
Synthesis of 6-bromo-7-fluoro-2-(trifluoromethyl)quinolin-4(1H)-one (17)
A mixture of 4-bromo-3-fluoroaniline (30 g, 0.157 moles, 1.0 equiv), ethyl 4,4,4-trifluoro-3-oxobutanoate (34.88 mL, 0.236 moles, 1.5 equiv), and polyphosphoric acid (150 g, 5 times w/w, w.r.t SM) was stirred using a mechanical stirrer and heated to 140 °C for 2 h. LCMS confirmed the completion of the reaction. The reaction mass was allowed to cool to rt and then diluted with 10% aqueous sodium hydroxide (during this process, the first solid was formed, which further disappeared when pH 9 was obtained). Some insoluble materials were removed by filtration, and the clear solution was acidified with concentrated HCl (36%). Obtained solids were collected and crystallized from IPA yield of 24.47 g, percent yield 50%, off-white solid; M. Pt (290 °C); 1H NMR (400 MHz, DMSO-d6) δ 12.84 (s, NH, 1H), 8.49 (s, CH, 1H), 8.48–7.97 (s, CH, 1H), 7.09 (s, CH, 1H);13C NMR (400 MHz, DMSO) δ 160.47, 157.99, 134.44, 127.68, 125.21, 122.48, 119.74, 113.10, 109.30, 101.21; MS (m/z) calculated: 310.04, MS (m/z) observed: [M-H]+: 309.8.
Synthesis of 3-(2-(6-bromo-7-fluoro-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)-1-cyclopropylimidazolidine-2,4-dione (18)
To a solution of 6-bromo-7-fluoro-2-(trifluoromethyl) quinoline-4(1H)-one (24.0 g, 0.050 moles, 1.0 equiv) in DMF (480 mL, 20 v), K2CO3 (20.89 g, 0.151 moles, 3.0 equiv) and Et3N (21.08 mL, 0.151 moles, 3.0 equiv) were added followed by addition of 3-(2-bromomethyl)-1-cyclopropylimidazolidine-2,4-dione (24.90 g, 0.100 moles, 2.0 equiv) under N2 atmosphere. The whole reaction mass was stirred at 80 °C for 48 h. Ethyl acetate/water workup was performed. The organic layer was washed with a 15% brine solution to get rid of DMF. Finally, the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to obtain crude material, purified by column chromatography using 60–120 mesh silica and ethyl acetate/hexanes as an eluent. The yield obtained was 9.5 g, percent yield 26%, white solid; M.Pt (164 °C); 1H NMR (400 MHz, DMSO-d6) δ 8.53 (d, J = 8.00 Hz, CH, 1H), 8.08 (d, J = 12.00 Hz, CH, 1H), 7.48 (m, CH, 1H), 4.45–4.61 (m, CH2, 2H), 3.96 (s, CH2 (hydantoin), 2H), 3.92–3.90 (m, CH2, 2H), 2.61 (m, CH (cyclopropyl), 1H), 0.70 (s, CH2CH2(cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.3, 162.0, 159.1, 148.6 (d, J = 248.6 Hz), 156.5, 149.5 (q, J = 33.6 Hz), 147.4 (d, J = 12.3 Hz), 121.2 (q, J = 274.0 Hz), 174.0, 114.1 (d, J = 22.2 Hz), 110.8 (d, J = 23.9), 98.2, 67.3, 50.0, 37.0, 24.5, 5.2,; MS (m/z) calculated: 476.22, MS (m/z) observed: [M+H]+: 477.0.
General procedure for 1-cyclopropyl-3-(2-(7-fluoro-6-(1-methyl-1H-pyrazol-5-yl)-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl) imidazolidine-2,4-dione (19a–n)
To a solution of 3-(2-(6-bromo-7-fluoro-4-oxo-2-(trifluoromethyl)quinoline-1(4H)-yl)ethyl)-1-cyclopropyl imidazolidine-2,4-dione (250 mg, 0.0005 moles, 1.0 equiv) in DMF (2.5 mL, 10 v), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (131 mg, 0.0006 moles, 1.2 equiv) was added, followed by addition of K2CO3 (217 mg, 0.0015 moles, 3.0 equiv), and the mixture was purged with N2 for 5 min, and then Pd(dppf)Cl2.DCM (21 mg, 0.00002 moles, 0.05 equiv) was added to it and the reaction mass was purged with N2 for 2 min. Then, it was heated at 80 °C for 4 h. TLC confirmed the completion of the reaction. After completion of the reaction, ethyl acetate/water workup was performed. The organic layer was washed with a 15% brine solution (10 v × 3) to get rid of DMF. Finally, the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to obtain the crude material. The crude material was purified by column chromatography using 100–200 mesh silica and ethyl acetate/hexanes as an eluent.
1-cyclopropyl-3-(2-(7-fluoro-6-(1-methyl-1H-pyrazol-5-yl)-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)imidazolidine-2,4-dione (19a)
Off-white solid; M.Pt (173 °C);1H NMR (400 MHz,DMSO-d6) δ 8.32 (d, J = 8.00 Hz, CH, 1H), 8.07 (d, J = 8.00 Hz, CH, 1H), 7.61 (s, CH, 1H), 7.48 (s, CH, 1H), 6.57 (s, CH, 1H), 4.49–4.46 (m, CH2, 2H), 3.97–3.85 (m, CH3 (pyrazole methyl), CH2CH2, 7H), 2.54 (m, CH (cyclopropyl), 1H), 0.59–0.57 (m, (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.3, 163.0, 160.3 (d, J = 257.7 Hz), 156.5, 149.7 (q, J = 33.7 Hz), 148.2 (d, J = 13.4 Hz), 138.2, 135.2, 125.6 (d, J = 3.8 Hz), 121.6 (q, J = 273.8 Hz) 120.4 (d, J = 19.0 Hz), 119.8, 118.2, 113.6, 108.2, 98.0, 67.3, 50.0, 37.4, 37.3. 37.1, 24.4, 5.0; MS (m/z) [M+H]+: 478.
1-cyclopropyl-3-(2-(7-fluoro-4-oxo-6-(pyridin-3-yl)-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl) imidazolidine-2,4-dione (19b)
Off-white solid; M.Pt (165 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.91 (s, CH, 1H), 8.70 (d, J = 4.00 Hz, CH, 1H), 8.45 (d, J = 8.00 Hz, CH, 1H), 8.14 (d, J = 8.00 Hz, CH, 1H), 8.05 (d, J = 8.00 Hz, CH, 1H), 7.61 (d, J = 8.00 Hz, CH, 1H), 7.47 (s, CH, 1H), 4.48 (s, CH2 (hydantoin ring), 2H), 3.93–3.91 (m, CH2CH2, 4H), 2.66 (m, CH (cyclopropyl), 1H), 0.59 (s, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.3, 162.9, 160.8 (d, J = 251.1 Hz), 157.8, 149.6 (q, J = 3.4 Hz), 148.0 (d, J = 13.4 Hz), 136.5 (d, J = 2.9 Hz), 130.9, 127.5 (d, J = 17.6 Hz), 124.8, 123.7, 121.2 (d, J = 274.0 Hz), 118.6, 113.6, (d, J = 22.0 Hz), 97.8, 67.2, 50.0, 37.1, 24.5, 5.0,; MS (m/z) [M+H]+: 474.7.
1-cyclopropyl-3-(2-(7-fluoro-4-oxo-6-(prop-1-en-2-yl)-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl) imidazolidine-2,4-dione (19c)
White solid; M.Pt (135 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.26 (d, J = 8.00 Hz, CH, 1H), 7.87 (d, J = 12.00 Hz, CH, 1H), 7.40 (s, CH, 1H), 5.48 (s, CH2, 2H), 4.46–4.44 (m, CH2, 2H), 3.94 (s, CH2, 2H), 3.91 (s, CH2, 2H), 2.61–2.60 (m, CH (cyclopropyl), 1H), 2.25 (s, CH3, 3H), 0.67 (s, CH2CH2 (cyclopropyl), 4H) ; 13C NMR (400 MHz, DMSO) δ 170.2, 162.2, 161.3 (d, J = 252.3 Hz), 156.4, 149.0 (q, J = 33.5 Hz), 147.5 (d, J = 13.5 Hz), 138.2, 131.5 (d, J = 16.6 Hz), 122.9 (d, J = 5.7 Hz), 121.3 (d, J = 273.9 Hz), 118.9 (d, J = 5.8 Hz), 118.1, 113.2 (d, J = 23.0 Hz), 97.5, 67.1, 50.0, 37.1, 24.5, 22.7, 5.1; MS (m/z) [M+H]+: 437.9.
tert-butyl-4-(1-(2-(3-cyclopropyl-2,5-dioxoimidazolidin-1-yl)ethyl)-7-fluoro-4-oxo-2-(trifluoromethyl)-1,4-dihydroquinolin-6-yl)-5,6-dihydropyridine-1(2H)-carboxylate (19d)
Off-white solid; M.Pt (175 °C);1H NMR (400 MHz,DMSO-d6) δ 8.22 (d, J = 8.00 Hz, CH, 1H), 7.87 (d, J = 12.00 Hz, CH, 1H), 7.39 (s, CH, 1H), 6.27 (m, CH, 1H), 4.45 (s, CH2, 2H), 4.07 (s, CH2, 2H), 3.95 (s, CH2, 2H), 3.91–3.90 (m, CH2, 2H), 3.60 (s, CH2, 2H), 2.60 (m, CH2 (1,2,3,6-tetrahydropyridine), CH (cyclopropyl), 3H), 1.48 (s, t-butyl, 9H), 0.67–0.66 (m, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.2, 162.6, 161.1 (d, J = 251.7 Hz), 156.2, 153.8, 148.5 (q, J = 33.8 Hz), 147.3 (d, J = 13.6), 130.9 (d, J = 16.2 Hz), 130.0, 122.2 (d, J = 5.5 Hz), 120.5 (q, J = 273.7 Hz), 119.9, 118.2, 113.2 (d, J = 22.7 Hz), 97.5, 98.9, 67.0, 50.0, 37.1, 27.9, 24.5, 5.1,; MS (m/z) [M+H]+: 578.9.
tert-butyl4-(4-(1-(2-(3-cyclopropyl-2,5-dioxoimidazolidin-1-yl)ethyl)-7-fluoro-4-oxo-2-(trifluoromethyl)-1,4-dihydroquinolin-6-yl)phenyl) piperazine-1-carboxylate (19e)
Light-yellow solid; M.Pt (168 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.33 (d, J = 8.00 Hz, CH2, 1H), 7.94 (d, J = 12.00 Hz, CH, 1H), 7.62 (d, J = 8 Hz, CH2, 2H), 7.40 (s, CH, 1H), 7.11 (d, J = 12 Hz, CH2, 2H), 4.47 (s, CH2, 2H), 3.94–3.91 (m, 4H), 3.49 (s, CH2NCH2 (piperazine), 4H), 3.23 (s, CH2NCH2 (piperazine), 4H), 2.58 (s, CH (cyclopropyl), 1H), 1.43 (s, t-butyl, 9H), 0.64–0.61 (m, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.2, 162.6, 161.1 (d, J = 251.2 Hz), 156.4, 153.8, 150.7, 148.5 (q, J = 33.7 Hz), 147.2 (d, J = 13.5 Hz), 130.6 (d, J = 16.5 Hz), 129.8,124.1, 123.0, 121.4, (q, J = 273.9 Hz), 118.6, 115.3, 113.4 (d, J = 22.8 Hz), 97.4, 79.0, 67.0, 64.9, 50.9, 47.6, 37.2, 28.0, 24.5, 5.1,; MS (m/z) [M+H]+: 657.9.
1-cyclopropyl-3-(2-(6-(2,6-dimethoxyphenyl)-7-fluoro-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)imidazolidine-2,4-dione (19f)
Off-white solid; M.Pt (188 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.05 (d, J = 8.00 Hz, CH, 1H), 7.88 (d, J = 12.00 Hz, CH, 1H), 7.45–7.39 (m, 2H), 6.82 (d, J = 8.00 Hz, 2H), 4.46–4.45 (m, CH2 (hydantoin ring), 2H), 3.89–3.86 (m, 4H), 3.69 (s, OCH3, 6H), 2.58 (m, CH (cyclopropyl), 1H), 0.56 (s, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.0, 162.6, 161.9 (d, J = 249.3 Hz), 157.6, 156.3, 148.8 (q, J = 33.4 Hz), 148.0 (d, J = 13.3 Hz), 130.5, 126.4, (d, J = 5.1 Hz), 125.1 (d, J = 21.0 Hz), 121.3 (q, J = 273.8 Hz), 118.0, 112.6 (d, J = 22.2 Hz), 111.3, 104.2, 97.3, 66.7, 55.8, 49.7, 37.0, 24.3, 5.0; MS (m/z) [M+H]+: 534.2.
1-cyclopropyl-3-(2-(6-(2,5-dichlorophenyl)-7-fluoro-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)imidazolidine-2,4-dione (19g)
White solid; M.Pt (154 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.22 (d, J = 8.00 Hz, CH, 1H), 8.05–8.02 (m, CH, 1H), 7.72–7.69 (m, CH CH, 2H), 7.65–7.60 (m, CH, 1H), 7.48 (s, CH, 1H), 4.47–4.46 (m, CH2 (hydantoin), 2H), 3.91–3.85 (m, CH2CH2, 4H), 2.53 (m, CH (cyclopropyl),1H), 0.56 (s, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.0, 163.0, 160.5 (d, J = 250.3 Hz), 156.4, 149.7 (q, J = 33.7 Hz), 148.2 (d, J = 13.3 Hz), 135.3, 132.0, 131.8, 131.6, 131.4, 127.7, (d, J = 19.8 Hz), 125.7, 121.2, (q, J = 274.1 Hz), 118.2, 113.0 (d, J = 21.7 Hz), 97.9, 67.0, 50.0, 37.0, 24.4, 5.0; MS (m/z) [M+H]+: 541.8.
1-cyclopropyl-3-(2-(7-fluoro-4-oxo-6-(thiophen-2-yl)-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)imidazolidine-2,4-dione (19h)
Off-white solid; M.Pt (193 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.65 (d, J = 8.00 Hz, CH, 1H), 8.01 (d, J = 12.00 Hz, CH, 1H), 7.92–7.91 (m, CH, 1H), 7.84–7.82 (m, CH, 1H), 7.42 (s, CH, 1H), 7.30 (t, J = 4.00 Hz, CH, 1H), 4.49–4.46 (m, CH2 (hydantoin), 2H), 3.97–3.94 (m, CH2CH2, 4H), 2.63–2.58 (m, CH (cyclopropyl), 1H), 0.66–0.60 (s, CH2CH2 (cyclopropyl),4H); 13C NMR (400 MHz, DMSO) δ 170.3, 162.6, 159.7 (d, J = 252.2 Hz), 156.5, 148.9 (q, J = 33.6 Hz), 147.0 (d, J = 13.7 Hz), 134.9, 128.6 (d, J = 5.07 Hz), 128.0, 127.7, 122.6, 121.4, 121.2 (q, J = 274.0 Hz), 118.6, 113.6 (d, J = 22.4 Hz), 97.6, 67.2, 49.9, 37.2, 24.6, 5.1; MS (m/z) [M+H]+: 480.2.
2-(1-(2-(3-cyclopropyl-2,5-dioxoimidazolidin-1-yl)ethyl)-7-fluoro-4-oxo-2-(trifluoromethyl)-1,4-dihydroquinolin-6-yl)benzaldehyde (19i)
White solid; M.pt (153 °C); 1H NMR (400 MHz,DMSO-d6) δ 9.93 (s, CHO,1H), 8.24 (d, J = 8.00 Hz, CH, 1H), 8.07 (d, J = 8.00 Hz, CH, 1H), 7.98 (d, J = 8.00 Hz, CH, 1H), 7.86 (d, J = 8.00 Hz, CH,1H), 7.77 (d, J = 8.00 Hz, CH, 1H), 7.59 (d, J = 8.00 Hz, CH, 1H), 7.48 (s, CH, 1H), 4.47–4.46 (m, CH2 (hydantoin), 2H), 3.89–3.86 (m, CH2CH2, 4H), 2.58 (m, CH (cyclopropyl), 1H), 0.54 (s, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 191.8, 170.2, 162.9, 160.8 (d, J = 248.7 Hz), 156.4, 149.3 (q, J = 33.6 Hz), 148.4 (d, J = 13.2 Hz), 135.8, 134.2, 133.8, 131.9, 130.1, 129.3 (d, J = 39.5 Hz), 128.9, 125.4 (d, J = 4.5 Hz), 121.3 (q, J = 274.0 Hz), 118.4, 112.6 (d, J = 22.3 Hz), 97.8, 67.0, 50.0, 37.0, 24.4, 5.0; MS (m/z) [M+H]+: 502.2.
3-(2-(6-(5-chloro-2-methoxyphenyl)-7-fluoro-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)-1-cyclopropylimidazolidine-2,4-dione (19j)
White solid; M.pt (126 °C);1H NMR (400 MHz,DMSO-d6) δ 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 8.00 Hz, CH, 1H), 7.93 (d, J = 8.00 Hz, CH, 1H), 7.56–7.55 (m, CH, 1H), 7.54–5.51 (m, CHCH, 2H), 7.22 (d, J = 8.00 Hz, CH, 1H), 4.47–4.45 (m, CH2 (hydantoin), 2H), 3.91 (s, CH2, 2H), 3.88 (s, CH2, 2H), 3.77 (s, OCH3, 3H), 2.54–2.53 (m, CH (cyclopropyl), 1H), 0.58- 0.56 (m, CH (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.1, 162.8, 161.2 (d, J = 251.0 Hz), 156.4, 155.7, 149.2 (q, J = 33.7 Hz), 148.2 (d, J = 13.2 Hz), 130.3, 129.8, 127.5 (d, J = 20.2 Hz), 125.5, 125.4, 125.3, 124.2, 121.3 (q, J = 274.0 Hz), 118.2, 113.1, 112.5 (d, J = 22.2 Hz), 97.6, 66.9, 56.0, 49.9, 37.0, 24.4, 5.0; MS (m/z) [M+H]+: 538.2.
3-(2-(6-(benzofuran-2-yl)-7-fluoro-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl)-1-cyclopropylimidazolidine-2,4-dione (19k)
Beige solid; M.pt (177 °C); 1H NMR (400 MHz,DMSO-d6) δ 8.83 (d, J = 8.00 Hz, CH, 1H), 8.07 (d, J = 12.00 Hz, CH, 1H), 7.79 (d, J = 8.00 Hz, CH, 1H), 7.71 (d, J = 8.00 Hz, CH, 1H), 7.56 (d, J = 4.00 Hz, CH, 1H), 7.50 (s, CH, 1H), 7.45 (t, J = 8.00 Hz, CH, 1H), 7.34 (t, J = 8.00 Hz, CH, 1H), 4.58–4.55 (m, CH2 (hydantoin), 2H), 3.98–3.95 (m, CH2CH2, 4H), 2.60–2.59 (m, CH (cyclopropyl), 1H), 00.65–0.64 (m, CH2 (cyclopropyl), 2H), 0.59–0.57 (m, CH2 (cyclopropyl), 2H); 13C NMR (400 MHz, DMSO) δ 170.1, 162.7, 159.7 (d, J = 254.5 Hz), 156.2, 153.9, 149.2 (q, J = 33.7 Hz), 148.2 (d, J = 13.2 Hz), 147.7 (d, J = 13.5 Hz), 128.4, 125.7, 123.3, 121.8, 121.2 (q, J = 273.7 Hz), 120.6, 119.6, 119.5, 118.4, 113.7 (d, J = 21.4 Hz), 111.0, 108.0 (d, J = 11.9 Hz), 97.8, 66.6, 50.0, 36.9, 24.4, 5.0; MS (m/z) [M+H]+: 514.2.
1-cyclopropyl-3-(2-(7-fluoro-6-(furan-2-yl)-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl) imidazolidine-2,4-dione (19l)
Off-white solid; M.pt (155 °C);1H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 8.00 Hz, CH, 1H), 8.01–7.98 (m, CHCH, 2H), 7.44 (s, CH, 1H), 7.11 (s, CH, 1H), 6.76 (s, CH, 1H), 4.57–4.52 (m, CH2 (hydantoin), 2H), 3.95–3.93 (m, CH2CH2, 4H), 2.61 (m, CH (cyclopropyl),1H), 0.66 (s, CH2CH2 (cyclopropyl), 4H), 13C NMR (400 MHz, DMSO) δ170.1, 162.6, 159.2 (d, J = 253.4 Hz), 156.3, 148.4 (q, J = 34.0 Hz), 147.1 (d, J = 13.4 Hz), 146.5 (d, J = 2.7 Hz), 144.4, 121.2 (q, J = 273.9 Hz), 120.1 (d, J = 15.7 Hz), 118.9 (d, J = 4.5 Hz), 118.5, 115.6, 113.6 (d, J = 21.4 Hz), 112.6, 112.0 (d, J = 10.4 Hz), 97.7, 66.7, 49.9, 37.0, 24.5, 5; MS (m/z) [M+H]+: 464.2.
3-(2-(6-(4-acetylphenyl)-7-fluoro-4-oxo-2-(trifluoromethyl)-1(4H)-yl)ethyl)-1-cyclo propyl imidazoli-dine-2,4-dione (19m)
Off-white solid; M.pt (176 °C); 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 8.00 Hz, CH, 1H), 8.12 (d, J = 8.00 Hz, CH, 2H), 8.03 (d, J = 12.00 Hz, CH, 1H), 7.85 (d, J = 8.00 Hz, CH, 2H), 7.46 (s, CH, 1H), 4.50–4.47 (m, CH2 (hydantoin), 2H), 3.94–3.90 (m, CH2CH2, 4H), 2.66 (s, COCH3, 3H), 2.59–2.54 (m, CH (cyclopropyl), 1H), 0.62–0.57 (m, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 198.1, 170.2, 163.0, 160.7 (d, J = 251.5 Hz), 156.4, 149.4 (q, J = 33.7 Hz), 148.0 (d, J = 13.2 Hz), 138.5, 136.4, 129.7, 129.5, 129.4, 124.9 (d, J = 4.5 Hz), 121.5 (q, J = 273.6 Hz), 118.5, 113.6, (d, J = 22.6 Hz), 67.2, 50.9, 37.1, 26.8, 24.5, 5.0; MS (m/z) [M+H]+: 516.2.
1-cyclopropyl-3-(2-(6-cyclopropyl-7-fluoro-4-oxo-2-(trifluoromethyl)quinolin-1(4H)-yl)ethyl) imidazolidine-2,4-dione (19n)
Off-white solid; M.pt (165 °C); 1H NMR (400 MHz,DMSO-d6) δ 7.84 (d, J = 8.00 Hz, CH, 1H), 7.80 (d, J = 8.00 Hz, CH, 1H), 7.33 (s, CH, 1H), 4.41–4.39 (m, CH2 (hydantoin) 2H), 3.96 (s, CH2, 2H), 3.92–3.90 (m, CH2, 2H), 2.65–2.61 (m, CH (cyclopropyl),1H), 2.22 (s, CH (cyclopropyl),1H), 1.13–1.11 (m, CH2 (cyclopropyl) 2H), 0.99 (s, CH2 (cyclopropyl), 2H), 0.70–0.68 (m, CH2CH2 (cyclopropyl), 4H); 13C NMR (400 MHz, DMSO) δ 170.3, 162.9 (d, J = J = 249.5 Hz), 162.0, 156.5, 147.8 (q, J = 33.5 Hz), 146.6 (d, J = 13.6 Hz, 133.9 (d, J = 17.9 Hz), 125.3 (q, J = 273.9 Hz), 118.8 (d, J = 5.7 Hz) 118.3, 111.9 (d, J = 21.9 Hz), 97.0, 67.0, 50.0, 37.2, 24.6, 9.0, 8.9, 8.8, 5.1; MS (m/z) [M+H]+: 438.2.
3.2. Antibacterial Activity
All the synthesized compounds (19a–n) were tested for antibacterial activity to determine the zone of inhibition against a Gram-positive bacterium (Staphylococcus aureus MTCC 96) and Gram-negative bacteria (Pseudomonas aeruginosa MTCC 441, Klebsiella pneumonia MTCC 109, and Escherichia coli MTCC 442).
3.3. Agar Well Plate Method
The newly synthesized compounds were diluted in DMSO with the required concentration for the bioassay. The bacterial cultures were grown on petri plates containing nutrient agar media (10 g beef extract, 2 g yeast extract, 5 g peptone, 5 g NaCl, and 15 g agar in 1 L of distilled water) at 37 °C. The autoclaved nutrient agar media was poured onto petri plates and allowed to solidify. After solidification, the culture was spread onto petri plates using an L-spreader and wells were created using a sterile 5 mm cork-borer [41,42,43,44].
With a micropipette, 20–40 µL of solution of the compounds was added into these wells. Ampicillin and chloramphenicol were used as positive controls to evaluate the potency of the tested compounds under the same conditions, with DMSO as the negative control. The plates were incubated at 37 °C for 15–20 h. The antibacterial activity was calculated by measuring the size of the zone of inhibition around each well using an antibiotic zone scale. Each experiment was performed thrice to obtain an average value. Minimum inhibitory concentration was determined for all the compounds using the agar dilution method by making different dilutions of the compounds with DMSO, such as 200, 100, 50, and 25 µg/mL. The greatest dilution showing at least 99% inhibition was taken as the minimum inhibitory concentration.
3.4. Molecular Modeling Studies
(a) Ligand and protein preparation
The structure of compound 19c was generated by MarvinSketch, which was optimized by minimizing the energy using the OPLS 2005 forcefield through the Ligprep module of Schrodinger Suite 2021 [45,46]. The ionization of the structure of compound 19c was retained in the original state and the optimized structure was saved in .sdf format for docking procedures. The co-crystallized structure of DNA gyrase (topoisomerase II) complexed with the 4,5’-bithiazole derivative, having the resolution of 1.50Å, was retrieved from the Protein Data Bank (PDB) with the accession number 4DUH [47,48,49,50]. The structure was downloaded in .pdb format and was further prepared using the Protein Preparation Wizard of Schrodinger Suite. The protein was first pre-processed by assigning the bond orders and hydrogen, converting selenomethionines to methionines, creating zero-order bonds to metals and adding disulfide bonds. The missing side chains and loops were filled using the Prime Module of Schrodinger Suite. All the water molecules beyond 5Å were deleted, the protein structure was pre-processed, and hydrogen bonds were assigned, which was followed by energy minimization by the OPLS 2005 forcefield [45]. A Ramachandran plot was obtained after the preparation of protein to validate the protein preparation, which justified that all the amino acids were in a favorable region. The final structure obtained was saved in .pdb format.
(b) Grid generation and validation of docking protocol
The co-crystallized ligand 4,5’-bithiazole derivative (accession number RLI) was downloaded from the PDB (PDB: 4DUH) in .sdf format and prepared in a similar manner as compound 19c by using the Ligprep module of Schrodinger Suite. The gird at the binding site of RLI was generated using the Receptor Grid Generation module of the Schrodinger Suite, which was defined by simply picking any atom of RLI within the active site that defined the grid coordinates as X: 2.47, Y: 2.71, and Z: 37.56. Rigid docking was performed with compound RLI at the defined binding site of DNA gyrase and root mean square deviation was computed using the Superposition tool of Schrodinger Suite, which was in a very acceptable range (RMSD: 0.0968Å), validating our docking protocol.
(c) Induced fit docking of compound 19c
Induced fit docking (IFD) of the prepared structure of compound 19c was performed to streamline its binding pattern within the defined binding site of DNA gyrase by using the extra-precision method in Schrodinger Suite. The twenty best poses were generated by induced fit docking, among which the best ten were selected to obtain the Interaction Fingerprint. Careful examination of the interactions and dock scores amongst these poses helped us to select the best pose, with a dock score of −10.091 (second pose in Interaction Fingerprint: Figure 4A), to explain the interactions of compound 19c with DNA gyrase.
4. Conclusions
In this study, trifluoromethylated quinolones tethered with hydantoin were designed, synthesized, and characterized using 1H and 13CNMR. All the synthesized hybrid molecules were evaluated for their antibacterial activity against one Gram-positive bacterial strain (Staphylococcus aureus MTCC 96) and three different Gram-negative bacterial strains (Pseudomonas aeruginosa MTCC 441, Klebsiella pneumonia MTCC 109, and Escherichia coli MTCC 442). Among all the hybrids, compound 19c became apparent as the most potent hybrid with the highest activity against S. aureus and K. pneumoniae, comparable to that of the standard drug chloramphenicol (MIC 50 µg/mL). Various binding interactions of 19c with the active sites of DNA gyrase of Staphylococcus aureus were run to explain their potential in blocking DNA gyrase. Thus, overall, the study concluded that these hybrid molecules could act as a lead for further development of antibacterial agents.
Author Contributions
Conceptualization, T.S.C. and A.A.; methodology, A.M.; software, H.S. and D.K.A.; validation, T.S.C., A.A. and A.S.; formal analysis, A.M.; investigation, A.M. and H.S.; resources, H.S.; data curation, A.M.; writing—original draft preparation, A.M. and A.A.; writing—review and editing, D.K.A., A.A. and T.S.C.; visualization, A.M. and A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge Guru Nanak Dev University Amritsar for analytical support and The NorthCap University for providing a research work and literature survey facility.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chu, D.T.W.; Plattner, J.J.; Katz, L.J. New directions in Antibacterial Research. Med.Chem. 1996, 39, 3853–3874. [Google Scholar] [CrossRef]
- Beovic, B. The issue of an antimicrobial resistance in human medicine. Int. J. Food Microbiol. 2006, 112, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.; Hunter, P.A.J. Antibiotic resistance-Action to promote new technologies. Antimicrob. Chemother. 2006, 58, i3–i22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suree, N.; Jung, M.E.; Clubb, R.T. Recent advances toward new anti-infective agents that inhibit cell surface protein anchoring in staphylococcus aureus and other gram positive pathogens. Mini-Rev. Med.Chem. 2007, 7, 991–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitscher, L.A.; Pillai, S.P.; Gentry, E.J.; Shankel, D.M. Mutiple drug resistance. Med. Res. Rev. 1999, 19, 477–496. [Google Scholar] [CrossRef]
- Nikaido, H.; Zgurskaya, H.I. Antibiotic efflux mechanism. Curr. Opin. Infect. Dis. 1999, 12, 529–536. [Google Scholar] [CrossRef]
- WHO. 2021. Available online: https://www.who.int/news-room/detail/29-04-2019-new-report-calls-for-urgent-action-to-avert-antibacterial-resistance-crisis (accessed on 21 September 2021).
- Marepu, N.; Yeturu, S.; Pal, M. 1,2,3-Triazole fused with pyridine/pyrimidine as new template for antimicrobial agents: Regioselective synthesis and identification of potent N-heteroarenes. Bioorg. Med. Chem. Lett. 2018, 28, 3302–3306. [Google Scholar] [CrossRef]
- El-Gohary, N.-S.; Shaaban, M.I. Design, synthesis, antimicrobial, antiquorum-sensing and antitumor evaluation of new series of pyrazolopyridine derivatives. Eur. J. Med. Chem. 2018, 157, 729–742. [Google Scholar] [CrossRef]
- Gao, F.; Yang, H.; Lu, T.; Chen, Z.; Ma, L.; Xu, Z.; Schaffer, P.; Lu, G. Design, synthesis and anti-mycobacterial activity evaluation of benzofuran-isatin hybrids. Eur. J. Med. Chem. 2018, 159, 277–281. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Wang, X.; Wang, Y.; Tang, W.-J.; Shi, J.-B.; Liu, X.-H. Novel curcumin analogue hybrids: Synthesis and anticancer activity. Eur. J. Med. Chem 2018, 156, 493–509. [Google Scholar] [CrossRef]
- Maestro, A.; Martín-Encinas, E.; Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Vicario, J.; Palacios, F. Synthesis of novel antiproliferative hybrid bis-(3-indolyl)methane phosphonate derivatives. Eur. J. Med. Chem. 2018, 158, 874–883. [Google Scholar] [CrossRef]
- Chopra, R.; Chibale, K.; Singh, K. Pyrimidine-chloroquinoline hybrids: Synthesis and antiplasmodial activity. Eur. J. Med. Chem. 2018, 148, 39–53. [Google Scholar] [CrossRef]
- Mott, B.T.; Cheng, K.C.C.; Guha, R.; Kommer, V.P.; Williams, D.L.; Vermeire, J.J.; Cappello, M.; Maloney, D.J.; Rai, G.; Jadhav, A.; et al. A furoxan-amodiaquine hybrid as a potential therapeutic for three parasitic diseases. MedChemComm 2012, 3, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Saini, A.; Kumar, S.; Raj, R.; Chowdhary, S.; Gendrot, M.; Mosnier, J.; Fonta, I.; Pradines, B.; Kumar, V. Synthesis and antiplasmodial evaluation of 1H-1, 2, 3-triazole grafted 4-aminoquinoline-benzoxaborole hybrids and benzoxaborole analogues. Bioorg. Chem. 2021, 109, 104733. [Google Scholar] [CrossRef]
- King, D.E.; Malone, R.; Lilley, S.H. New classification and update on the quinolone antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar]
- Ryu, C.K.; Sun, Y.J.; Shim, J.Y.; You, H.J.; Choi, K.U.; Lee, H. Synthesis and antifungal activity of 6,7-bis-[S-(aryl)thio]-5,8-quinolinediones. Arch. Pharm. Res. 2002, 25, 795–800. [Google Scholar] [CrossRef]
- Musiol, R.; Jamilek, J.; Buchta, V.; Silva, L.; Niedbala, H.; Podeszwa, B.; Palka, A.; Maniecka, K.M.; Oleksyn, B.; Polanski, J. Antifungal properties of new series of quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 3592–3598. [Google Scholar] [CrossRef]
- Desai, U.; Mitragotri, S.; Thopate, T.; Pore, D.; Wadgaonkarb, P. A highly efficient synthesis of trisubstitutedquinolines using sodium hydrogensulfate on silica gel as a reusable catalyst. Arkivoc 2006, 15, 198–204. [Google Scholar]
- Nilsen, A.; LaCrue, A.N.; White, K.L.; Forquer, I.P.; Cross, R.M.; Marfurt, J.; Mather, M.W.; Delves, M.J.; Shackleford, D.M.; Saenz, F.E.; et al. Quinolone-3-diarylethers: A new class of antimalarial drug. Sci Trans. Med. 2013, 5, 177ra37. [Google Scholar] [CrossRef] [Green Version]
- Vu, A.T.; Cohn, S.T.; Manas, E.S.; Harris, H.A.; Mewshaw, R.E. ERβ ligands. Part 4: Synthesis and structure–activity relationships of a series of 2-phenylquinoline derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4520–4525. [Google Scholar] [CrossRef]
- Gogoi, S.; Shekarrao, K.L.; Duarah, A.; Bora, T.C.; Boruah, R.C. A microwave promoted solvent-free approach to steroidal quinolines and their in vitro evaluation for antimicrobial activities. Steriods 2012, 77, 1438–1445. [Google Scholar] [CrossRef]
- Ranu, B.C. Indium Metal and Its Halides in Organic Synthesis: Microreview. Eur. J. Org. Chem. 2000, 2000, 2347–2356. [Google Scholar] [CrossRef]
- Prajapati, S.M.; Patel, K.D.; Vekariya, R.H.; Panchal, S.N.; Patel, H.D. Recent advances in the synthesis of quinolines: A review. RSC Adv. 2014, 4, 24463–24467. [Google Scholar] [CrossRef]
- Amii, H.; KishiKawa, Y.; Uneyama, K. Rh(I)-Catalyzed Coupling Cyclization of N-Aryl Trifluoroacetimidoyl Chlorides with Alkynes: One-Pot Synthesis of Fluorinated Quinolines. Org. Lett. 2001, 3, 1109–1112. [Google Scholar] [CrossRef]
- Mahajan, A.; Chudawat, T.S. Review on the role of the metal catalyst for synthesis of pharmacologically important quinoline substrate. Mini Rev. Org. Chem. 2019, 16, 631–652. [Google Scholar] [CrossRef]
- McNaughton, B.R.; Miller, B.L. A mild and efficient one-step synthesis of quinolines. Org. Lett. 2003, 5, 4257–4259. [Google Scholar] [CrossRef]
- Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Recent advances in the synthesis of hydantoins: The state of the art of a valuable scaffold. Chem. Rev. 2017, 117, 13757–13809. [Google Scholar] [CrossRef]
- Lopez, L.I.L.; Loera, D.D.; Avalos, E.R.; Galindo, A.S. Green synthesis of hydantoins and its derivatives. Mini Rev. Org. Chem. 2020, 17, 176–184. [Google Scholar] [CrossRef]
- Fujisaki, F.; Toyofuku, K.; Egami, M.; Ishida, S.; Nakamoto, N.; Kashige, N.; Miake, F.; Sumoto, K. Antibacterial activity of some 5- dialkylaminomethylhydantoins and related derivatives. Chem. Pharm.Bull. 2013, 61, 1090–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehrani, K.H.M.E.; Hashemi, M.; Hassan, M.; Kobarfard, F.; Mohebbi, S. Synthesis and antibacterial activity of Schiff bases of 5-substituted isatins. Chin. Chem. Lett. 2016, 27, 221–225. [Google Scholar] [CrossRef]
- Bapna, M.; Parashar, B.; Sharma, V.K.; Chouhan, L.S. Microwave-Assisted synthesis of some novel and potent antibacterial and antifungal compounds with biological screening. Med. Chem. Res. 2012, 7, 1098–1106. [Google Scholar] [CrossRef]
- Alaa, A.B.; Hanin, A.B.; Safwat, A.A.; Reem, M.D.; Sameh, S.E. New bromoindole alkaloid isolated from the marine spongeHyrtios erectus. Heterocycles 2018, 96, 749–756. [Google Scholar]
- Nagai, T.; Nishioka, G.; Koyama, M.; Ando, A.; Miki, T.; Kumadaki, I. Reactions of trifluoromethyl ketones. IX. Investigation of the steric effect of a trifluoromethyl group based on the stereochemistry on the dehydration of trifluoromethyl homoallyl alcohols. J. Fluorine Chem. 1992, 57, 229–237. [Google Scholar] [CrossRef]
- Bravo, P.; Dillido, D.; Resnati, G. An efficient entry to perfluoroalkyl substituted azoles starting from β-perfluoroalkyl-β-dicarbonyl compounds. Tetrahedron 1994, 50, 8827–8836. [Google Scholar] [CrossRef]
- Gregory, T.P.; Darlene, C.D.; David, M.S. Structure-Activity Relationships for the Action of Dihydropyrazole Insecticides on Mouse Brain Sodium Channels Pestic. Biochem. Phys. 1998, 60, 177–185. [Google Scholar]
- Kucukguzel, S.G.; Rollas, S.; Erdeniz, H.; Kiranz, A.C.; Ekinci, M.; Vidin, A. Synthesis, characterization and pharmacological properties of some 4-arylhydrazono-2-pyrazoline-5-one derivatives obtained from heterocyclic amines. Eur. J. Med. Chem. 2000, 35, 761–771. [Google Scholar] [CrossRef]
- Marull, M.; Schlosser, M. Selective and efficient structural elaboration of 2-(trifluoromethyl)quinolinones. Eur. J. Org. Chem. 2003, 8, 1576–1588. [Google Scholar] [CrossRef]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. Fundam.Mol. Mech. Mutagen. 2007, 623, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Rizk, O.H.; Bekhit, M.G.; Hazzaa, A.A.B.; El-Khawass, E.M.; Abdelwahab, I.A. Synthesis, antibacterial evaluation and DNA gyrase inhibition profile of some newquinoline hybrids. Arch. Pharm. 2019, 352, 1900086. [Google Scholar] [CrossRef]
- Omar, A.M.; Alswah, M.; Ahmed, H.E.A.; Bayoumi, A.H.; El-Gamal, K.M.; El-Morsy, A.; Ghiaty, A.; Afifi, T.H.; Sherbiny, F.F.; Mohammed, A.S.; et al. Antimicrobial screening and pharmacokinetic profiling of novel phenyl-[1,2,4]triazolo[4,3-a] quinoxaline analogues targeting DHFR and E. coli DNA gyrase B. Bioorg. Chem. 2020, 96, 103656. [Google Scholar] [CrossRef]
- Hooper, D.C.; Jacoby, G.A. Topoisomerase inhibitors: Fluoroquinolone mechanisms of action and resistance. Perspect. Med. 2016, 6, a025320. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Valgas, C.; Souza, S.M.D.; Smânia, E.F.; Smânia, A. Screening methods to determine antibacterial activity of natural products. Braz. J. Microbiol. 2007, 38, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Arya, A.; Gupta, K.; Chundawat, T.S.; Vaya, D. Biogenic synthesis of copper and silver nanoparticles using green alga Botryococcus braunii and its antimicrbial activity. Bioinorg. Chem. Appl. 2018, 2018, 7879403. [Google Scholar] [CrossRef] [Green Version]
- DeAlba-Montero, I.; Guajardo-Pacheco, J.; Morales-Sánchez, E.; Araujo-Martínez, R.; Loredo-Becerra, G.M.; Martínez-Castañón, G.A.; Compeán Jasso, M.E. Antimicrobial properties of copper nanoparticles and amino acid chelated copper nanoparticles produced by using a soya extract. Bioinorg. Chem. Appl. 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665. [Google Scholar] [CrossRef] [Green Version]
- LigPrep, Version 2.3; Schrodinger, LLC.: New York, NY, USA, 2009.
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-Based Discovery of Substituted 4,5′-Bithiazoles as Novel DNA Gyrase Inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).