Abstract
The very high capture cross-section of (epi)thermal neutrons by the boron isotope 10B makes elemental boron and its compounds and composites prospective for serving as materials intensively interacting with neutron irradiation. In their nanostructured form, boron-rich materials reveal properties that improve their radiation-performance characteristics. In this regard, new technologies have been proposed for the synthesis of nanocomposites with matrices of boron carbide B4C and hexagonal boron nitride h-BN. For the first time, boron carbide-tungsten and hexagonal boron nitride–(iron,magnetite) composites were obtained, respectively, in the form of layered/sandwich structures of components B4C and W and h-BN nanopowders coated/intercalated with magnetic nanoclusters of iron Fe or magnetite Fe3O4. Studying of their chemical/phase composition, structure/morphology, and some other properties leads to the conclusion that the developed B4C–W and h-BN–(Fe,Fe3O4) composites would be useful for solving important problems of boron-based neutron shielding and BNCT (Boron Neutron Capture Therapy), such as attenuating the gamma-radiation accompanying the absorption of neutrons by 10B nuclei and targeted delivery of 10B nuclei, as BNCT therapeutic agents, to tumor tissues using control by an external magnetic field, respectively.
1. Introduction
Because of the high capture cross section of (epi)thermal neutrons by the boron 10B isotope, boron and its compounds and composites are widely used for materials intensively interacting with neutron radiation. In the fine-dispersive form, they reveal additional properties, improving performance characteristics. In particular, it has been reported on the preparation methods of two important boron-containing fine-dispersive composites useful in neutron shielding and BNCT (Boron Neutron Capture Therapy), respectively [1,2].
Boron carbide–tungsten B4C–W thin-layered sandwich structures are known [3,4,5] as effective shield composite materials, in which low- and high-Z atoms (Z is the atomic number), B and W, serve as effective absorbers for, respectively, (epi)thermal neutrons and secondary gamma-quanta accompanying the neutrons captured by 10B nuclei. W- and B-based thin film materials, including B4C (as well as WC and WB3), display very high hardness [6]. To expand the room-temperature superior mechanical properties of W foils, it was proposed [7] to fabricate tungsten-containing metallic laminate composites.
The effect of neutron irradiation on W foil was investigated to determine the resulting DBTT (Ductile to Brittle Transition Temperature) shift. Physical properties, processing techniques, and applications of high-operating temperature (>1200 °C) materials with multi-layered ceramic/carbon, ceramic, and metal structures were highlighted in [8].
These composites can have a graceful failure mode and higher toughness as compared toparticle-reinforced ceramic composites and, as multi-layered shields, provide better shielding efficacy than single-layered ones. Work [9] aimed to describe the development of an online platform to calculate (in the energy range of 0.015–15 MeV) the 36 GSPs (Gamma Shielding Parameters), which are required to investigate the materials gamma-ray shielding.
The influence of interface roughness on the reflectivity of B4C–W multilayers varying with bi-layer number N was specially investigated in [10]. For B4C–W multilayers with the same design period thickness of 2.5 nm, a real-structure model was used to calculate the variation of reflectivities with N = 50, 100, 150, and 200. These multilayers were fabricated by a DC (Direct Current) magnetron sputtering system. Their reflectivity and scattering intensity were measured by the XRD (X-ray Diffraction) method. The results indicate that reflectivity is a function of its bi-layer number, and interface roughness slightly increases from layer to layer during multilayer growth.
As for the hexagonal boron nitride h-BN nanopowders added with iron Fe ferromagnetic or its oxide Fe3O4 (magnetite) ferrimagnetic clusters, they were proposed [11] as prospective materials providing a magnetic field controlled delivery of 10B isotopes in BNCT. Another recent review [12] also emphasizes that BNCT is a high-LET (Linear Energy Transfer) therapy that facilitates tumor-directed radiation delivery of boron while largely sparing adjacent normal tissues through the targeting of boron compounds to tumor cells. Given this, developing novel boron agents with high selectivity, ease of delivery, and large boron payloads remains an area of active investigation.
Below, we briefly list the available literature on obtaining methods and some properties of B4C–W and h-BN–(Fe,Fe3O4)composites.
Tungsten layer can be obtained by WO3 oxide layer formation using the spin coating method to reduce it to α-W in a hydrogen atmosphere at 600–800 °C [13]. For the purpose of obtaining B4C–W layered composites, the liquid charge method previously developed [14,15,16,17,18,19] can be modified for multi-component B4C-matrix ceramics containing W compounds.
Due to the structural similarity between few-layered h-BN and graphene, they can reveal similar chemical properties, in particular their ability to be intercalated—see, e.g., [20,21]. Based on this analogy, the h-BN–Fe nanocomposites growth mechanism was proposed in [22].
Both of the technologies elaborated here are chemical synthesis routes, including the preparation of liquid charges from commercial precursor materials and their multi-stage thermal treatment.
The aim of the work is to develop methods for obtaining boron-containing nanocomposite materials intensively interacting with neutron radiation, namely, boron carbide-tungsten sandwich structures B4C–W and hexagonal boron nitride nanopowders doped with iron or magnetite magnetic clusters h-BN–(Fe,Fe3O4).
Boron carbide B4C is one of the superhard materials; its hardness is surpassed only by diamond C and cubic boron nitride c-BN. At the same time, boron carbide is quite light, which makes it a material with the highest hardness-to-density ratio among constructive materials widely used in modern technologies. It is why pure boron carbide and especially B4C-matrix composites are promising for the manufacture of neutron shields. The phenomenon of gamma-radiation accompanying the absorption of neutrons by 10B nuclei is an important challenge for boron carbide-based neutron shields. Here we propose to solve this problem by developing layered boron carbide–tungsten B4C–W structures, taking advantage of the ability of tungsten, a heavy metal, to intensely absorb gamma-rays, as well as the strong W–B and W–C bonding in the formed boron and carbon borides, respectively.
As for hexagonal boron nitride (h-BN), it is a layered crystal, a structural analogue of graphite, prone to easy exfoliation. Due to the high boron content (every second atom is B), ultra-small average particle size, on-demand biodegradability in the human body, layered crystal structure allowing its intercalation by foreign functional nanoclusters, etc., nanopowder h-BN-matrix composites are promising for the delivery of therapeutic agents of BNCT—boron 10B isotopes. An important challenge when using h-BN-based delivery agents in BNCT is targeting them to tumors to avoid damage to healthy tissues from neutron irradiation. Here we propose to solve this problem by doping hexagonal boron nitride nanoparticles with magnetic nanoclusters of ferromagnetic iron Fe or ferrimagnetic magnetite Fe3O4 and taking advantage of the phenomenon of controlling the flow of magnetic particles in the human body by an external magnetic field.
From the above characteristics of B4C- and h-BN-matrices, it is clear that there is no point in using superhard B4C and easily exfoliating h-BN for delivering a therapeutic agent in the human body and as a structural material, respectively. Therefore, below we focus not on obtaining the compositions B4C–(Fe,Fe3O4) and h-BN–W, but on B4C–W and h-BN–(Fe,Fe3O4).
2. Experimental—Materials and Equipment
2.1. Materials
Amorphous boron 0.2–1 μm powder (Figure 1) with a purity of 99.5% and boric acid are used to obtain boron nitride. Boron carbide 1–7 μm powders are purchased from Alfa Aesar. Ammonia, ammonium chloride, and ammonium hydroxide are used as nitrogen sources. Pure hexagonal boron nitride (h-BN) 0.1–1.1 μm powder (Figure 2) is used as a precursor. Tungsten powder is used as well.
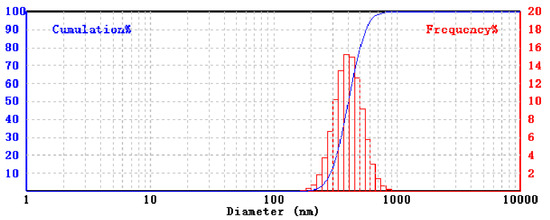
Figure 1.
Size distribution of precursor amorphous boron.
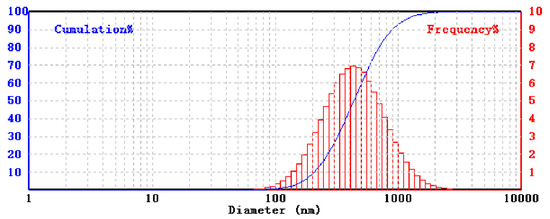
Figure 2.
Size distribution of precursor pure h-BN.
As iron sources are used, its compounds such as FeSO4∙7H2O, FeCl3∙6H2O, Fe(NO3)3∙9H2O, and Fe(CO)5. Reduction of iron compounds is performed with sodium borohydride (NaBH4) purchased from Sigma Aldrich. Organic compounds and polymers (carbohydrates, glucose, sucrose, cellulose, polyvinyl alcohol, polyvinylpyrrolidone, etc.) are used as carbon sources (for carbidization and as reducing agents), and pyrolysis produces activated amorphous carbon, so-called carbon black.
An inert environment is created in the reaction area using nitrogen and argon. The purity of chemical reagents and solvents used for synthesis reaches 99.00–99.99%. Reagents purchased from Sigma Aldrich are used without prior purification.
2.2. Technological Facilities
Thermal treatment (at temperatures of <1500 °C) of samples is implemented in the high-temperature vacuum furnace Kejia. High-temperature vacuum furnaces STG–100–17 (1700 °C) and Kejia are used for boron nitride synthesis and iron(III) chloride intercalation. In the same furnaces, metal oxides deposited on boron nitride are reduced by hydrogen flow.
To grind the powders, there is a planetary mill, the Pulverisette 7 Premium Line, with a grinding cup and balls made from WC–Co hard alloy.
Compaction of powder samples or simultaneous synthesis and compaction are carried out by using the SPS (Spark Plasma Sintering) equipment manufactured at the Georgian Technical University (Tbilisi, Georgia) with the ability to operate in DC, pulsed DC, and pulsed AC (Alternating Current) modes.
For the ultrasound treatment and homogenization of suspensions, an ultrasonic cleaner (45 kHz) and a JY92–IIDN Touch Screen Ultrasonic Homogenizer (20–25 kHz and 900 W) are used.
2.3. Measuring Apparatus
Particle sizes are determined by the photon correlation nanoparticle size analyzer Winner 802 DLS and the Malvern Instruments Mastersizer. The specific surface area of the composite powder is measured on the Micromeritics Gemini VII instrument.
The morphology and microstructure of the powders are studied with the SEM (Scanning Electron Microscope) JEOL–JSM 6510 LV (JEOL Ltd., Akishima, Tokyo, Japan) equipped with an energy dispersive analyzer, the Dispersive Micro-X-ray Spectral Analyzer X-MaxN (Oxford Instruments, Oxford, UK). XRD patterns are obtained with DRON–3M (CuKα, Ni filter, and 2°/min) and XZG–4 (CuKα and λ = 1.5418 Å) diffractometers. Powder particle sizes are determined by the Scherer method.
FTIR (Fourier Transform InfraRed) spectroscopy experiments are carried out on an Agilent Cary 630 (Agilent, Santa Clara, CA, USA) FTIR spectrometer with a 320-Cary FTIR Diamond ATR encompassing a spectrum range of 6300–350 cm−1. The measurements were performed on powder samples at room temperature in the presence of ambient air.
The VSM (Vibrating Sample Magnetometer) Lake Shore 7300 is used for the general characterization of the magnetic state of the received materials by measuring their room temperature magnetic properties, namely, magnetization in an external magnetic field up to 14 kG. Information on saturation and remnant magnetizations of these materials, Curie and Neel temperatures of coercive power, etc. can be obtained as well. The superparamagnetic properties of nanostructured materials doped with magnetic particles can also be determined.
3. Boron Carbide–Tungsten Layered Composites
3.1. Available Methods of Obtaining Tungsten Layers
Coating different types of surfaces with metallic tungsten is widely used in modern technologies such as microprocessors, DRA (Dynamic Random Access), flashmemories, image sensors, etc. First, it could be noted that semiconductors are coated with a thin layer of tungsten, which is mainly carried out by the CVD (Chemical Vapor Deposition) method, where SiH4, H2, and WF6 are used as precursors. It is confirmed that maintaining the vacuum chamber at pressures of about 20 to 760 Torr improves the tungsten deposition rate as well as the reflectivity of the formed W-surface [23].
Tungsten thin film was deposited on (100) silicon substrate by using the LPCVD (Low Pressure CVD) technique [24] (see also [25]). WF6 was used as a source gas for tungsten and SiH4 as a reducing gas for WF6. Tungsten thin film was deposited by either SiH4 or Si substrate reduction of WF6 under cold-wall conditions and by SiH4 reduction of WF6 under hot-wall conditions. It was shown that the nm W5Si3 tungsten silicide layer is formed at the tungsten–silicon interface only under gas-phase deposition, and the formation and growth of the WSi2 tungsten silicide layer commence at 700 °C for CVD films and at temperatures above 750 °C for films obtained with plasma-chemical deposition. This results in a drastic increase in their electrical resistance. Under optimal conditions, tungsten films of 8 × 10−6 Ω∙cm resistivity are produced.
Tungsten–IPA (Inorganic PeroxopolytungsticAcid) powder, which is soluble in water, was prepared [26] by dissolving metal tungsten in hydrogen peroxide and by evaporating residual solvent. Then, the solution of W–IPA was mixed with organic solvent, which was spin-coated on wafers. Metallic tungsten films were obtained after the reduction procedure. By selecting an appropriate organic solvent and irradiating it with UV (UltraViolet) light, the sheet resistance of such film could be remarkably reduced.
Tungsten is often obtained in the form of nanosized powders. Reduction of WO3 with the in situ produced hydrogen gas at 600 °C in work [27] is an approach for the production of W nanopowder. The results of XRD analysis showed that nanostructured α-W and β-W are present in the final product. DTA (Differential Thermal Analysis) showed that the β-W → α-W phase transition occurs around 435 °C. Powder suspensions or pastes of this type are then used to coat surfaces.
Metallic tungsten deposition technologies on various metal substrates were used [28] as well. Commonly applied metals include aluminum (Al), copper (Cu), and titanium (Ti).
Other deposition methods, such as ALD (Atomic Layer Deposition) and PNL (Pulsed Nucleation Layer) techniques, may also be used to form tungsten nucleation layers. Paper [29] describes the evolution of the reduction process of ultrafine tungsten powder in industrial conditions. Tungsten blue oxides were used for reduction in an industrial push furnace under a counter-current flow of hydrogen.The reduction was carried out according to the following scheme: WO2.9 → WO2.72 → WO2 → W.
Pure-W metallic films are often deposited by PVD (Physical Vapor Deposition), i.e., by sputtering or evaporation, or by CVD. The authors of [30] developed HWALD (HotWire-Assisted ALD) to form tungsten (W) with its filament heated up to 1700–2000 °C. Atomic hydrogen H was generated by the dissociation of molecular hydrogen H2, which reacted with WF6 at the substrate to deposit. In a typical case, CVD nucleation layers are deposited from WF6 and SiH4 with argon as a carrier gas.
With the aid of chemical vapor transport of WOx(OH)y, surface morphology transformed into rod-like, star-shaped cracking, florets, irregularly fibrous structures, and finally, spherical tungsten particles [13].
One of the practical and cheap methods is that metallic tungsten is coated on the ceramic material using the following technology. In this regard, the applicability of chemical solution deposition to fabricate metallic films on the inner surface of alumina tubules was established. The technique involves the preparation of tungsten oxide layers from a PTA (PeroxoTungsticAcid) precursor solution and their subsequent reduction to tungsten in the presence of hydrogen [31].
Sandwich structures can be obtained by the method described in the patent [32]. This invention discloses a boron carbide ceramic metallization preparation method. The method comprises the following steps: (1) Mixing powder, namely 10–40 wt.% of Mo, W and Ni according to their weights ratio, carrying out ball milling, and sieving through a 300-mesh sieve to obtain mixed metal powder; (2) Preparing paste by mixing the metal powder obtained in the step 1 with 5 wt.% ethylcellulose solution according to weight ratio of (100–120):30 to obtain metalized paste; (3) Printing the metalized paste obtained in the step 2 on the part, needing to be metalized, of a boron carbide ceramic part with the thickness of 20–30 μm, and drying; and (4) Metalizing by putting the boron carbide ceramic part dried in the step 3 into a metallization sintering furnace, introducing hydrogen, keeping the temperature at 1650–1680 °C for 30–35 min, keeping the dew point at 0–10 °C, and cooling along with the furnace. The formed Mo, W, and Ni proportioned metalized layer alloy material is close to a thermal expansion system of boron carbide; the bonding strength is high; and the metalized stress is small. Due to the sub-μm metal network structure, the metal is not prone to falling off after being brazed.
3.2. Developed Methods of Obtaining Boron Carbide–Tungsten Sandwich Structures
After summarizing the methods described in the Section 3.1, we have developed some novel routes for obtaining B4C–W sandwich-like composites.
3.2.1. Synthesis of Peroxotungstic Acid
PTA is synthesized in the following way. A total of 5.0 g of tungsten powder is slowly dissolved in 40 mL of a 20–25% H2O2 solution. The H2O2 solution is added to the tungsten powder in 3 portions during 2 h (note that their reaction is very exothermic!). Then, 15 mL of hydrogen peroxide solution is added to the reaction mixture again and stirred at room temperature for 5 h. The solution is left for 12 h and then filtered.
A yellowish, transparent solution is obtained. Excess H2O2 is decomposed using a platinum spiral, and the solution is evaporated under vacuum. An orange crystalline substance containing 85.4% WO3 is obtained. The productis heated at 600 °C for 4 h.
3.2.2. Tungsten Deposition on Boron Carbide Surface
The preparation of a coating solution to deposit W film on the B4C surface includes the following steps. A total of 1.25 g of previously synthesized PTA is dissolved in 20 mL of 0.5% hydroxyl ethylcellulose (HES 30000) aqueous solution, and 5 mL of ethyleneglycol is added to the resulting solution. PTA film deposition is carried out by the dipping technique on pre-sintered B4C cylinder-shaped samples (with a diameter of 12 and a height of 5–10 mm). The sample is lowered into the solution to a depth of 1–2 mm and left for 30 s, then dried at 120 °C. This operation is repeated 8–10 times.
The dried sample is fired in the air at 500 °C in a muffle furnace. As a result of this process, a WO3 layer is formed on the B4C surface. The resulting B4C–WO3 composite is placed in a tubular muffle filled with argon and heated to 500 °C. Then, the argon atmosphere is replaced with an Ar–H2 gas mixture stream containing up to 8–12 vol.% of hydrogen.
Reduction of WO3 is carried out at 600–800 °C for 2 h. The thickness of the resulting metallic tungsten film depends on the coating solution concentration and the number of dipping procedures.
3.2.3. Compaction of Boron Carbide and Tungsten Powders
The formation of a metallic tungsten layer of different thicknesses in a sandwich structure depends on the mass of the initial tungsten powder. The sandwich composite has been obtained as follows.
A 12 mm diameter graphite pressform lined with graphite foil is placed with a certain amount of commercial W powder and pressed with a punch. Then, boron carbide powder is added on top of the tungsten powder and is also pressed with a punch to smooth the surface of the deposited powder. This surface is covered with graphite foil, and a graphite punch is placed on top. The press form is placed in the SPS device, and after vacuuming the chamber, the powder is pressed at a pressure of 30–50 MPa and a temperature of 1500–1700 °C for 10 min of holding time.
Pulsed AC with a pulse duration of 5 μs and a pause of 1 μs is used to sinter these structures. The sample heating rate reaches 100–200 °C/min. The sample ware is cooled in a vacuum.
In this way, sandwich-like B4C–W composites are obtained, where the boundary layer between two components is well observable.
3.2.4. Compaction of Boron Carbide Powder with Tungsten Foil
On a 12 mm-diameter graphite press-form lined with graphite foil, a metallic tungsten disk of the same diameter with a thickness of 1–2 mm is placed, and then the boron carbide powder is poured on top. It is pressed with a punch to smooth the surface of the poured powder.
This surface is covered with graphite foil, and a graphite punch is placed on top. Sintering is carried out as described above. The obtained sandwich-like B4C–W composite samples are cooled in vacuum. They also clearly reveal the boundary layer between the B4C and W components.
3.3. Characterization of Boron Carbide–Tungsten Sandwich Structures
In this section, we give a characterization of the obtained boron carbide–tungsten layered composites, which includes their chemical and phase compositions, structure, and morphology.
As SEM and EDX (Energy Dispersive X-ray) studies of the sandwich fracture surface showed, on the contact surface of the B4C and W consolidated phases, there is diffusion of boron and tungsten atoms in opposite directions and the formation of the intermediate phase W2B5 (Figure 3 and Figure 4). Intermediate-phase W2B5 also plays the role of a binding (cementing) substance because there is no separation of phases when the samples are fractured and B4C and metallic W remain firmly connected. The formation of the intermediate phase is well observed when the contact area is enlarged (Figure 3b).

Figure 3.
SEM images of composite B4C–W obtained by SPS from B4C and W powders at 1600 °C: fracture surfaces (a,b) with different magnifications.

Figure 4.
EDX mapping of all elements (a) and separately B (b), C (c), and W (d) distributions in B4C and W phases contact regions.
It seems interesting that in the XDR pattern of the boron carbide substrate with the removed tungsten coat (Figure 5), in addition to the main phase B4C (lattice parameters: a = 5.400011, b = 5.400011, c = 11.595017 Å; space group: trigonal, R–3m), the tungsten boride W2B5 phase (lattice parameters: a = 3.0171, c = 15.7082 Å; space group: hexagonal, P63/mmc) is detected as well. As for trace phases, like chromium carbide Cr3C2 (lattice parameters: a = 2.727, c = 2.538 Å; space group: hexagonal, Pnma), they may be related to the sample polishing process.
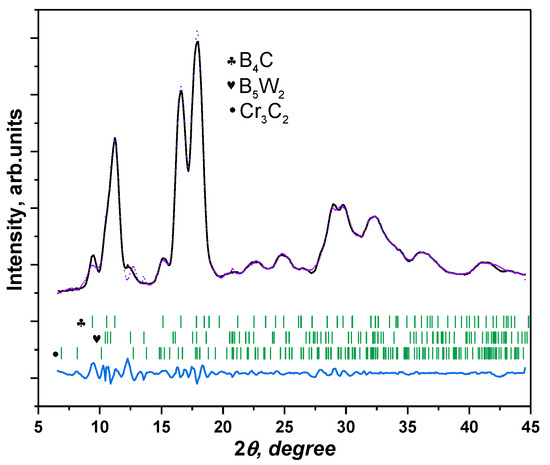
Figure 5.
XRD pattern of pure (with a removed tungsten coat) boron carbide. Detected phases: B4C, W2B5, and Cr3C2.
The thickness of the layers can be adjusted within certain limits. After removing tungsten and its boride layer from the sample sintered at 1600 °C, the relative density of boron carbide is determined to be 87–92% of the theoretical value. The existence of an intermediate phase is confirmed by a special experiment that makes it possible to selectively remove metallic tungsten with a 30% solution of hydrogen peroxide. The XRD study of the cleaned surface (Figure 6) unambiguously confirms its presence and also the removal of metallic tungsten from the sandwich surface. XRD patterns show the presence of only traces of the metallic W phase.
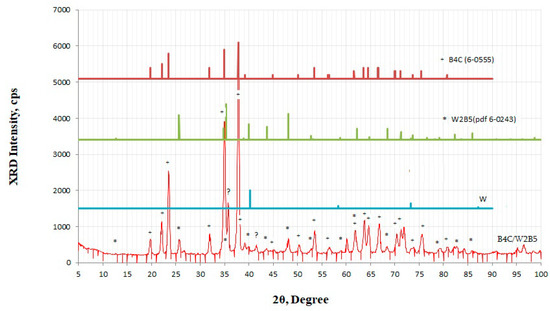
Figure 6.
XRD pattern of B4C–W2B5 composite obtained from B4C–W2B5–W sandwich composite after selectively removing the metallic tungsten layer by treatment with a 30% solution of hydrogen peroxide. Peaks corresponding to B4C and W2B5 component- and W trace-phases are shown separately.
3.4. Further Analysis of Boron Carbide–Tungsten Composites
Results obtained on boron carbide–tungsten composites can be further analyzed by taking into account information on B–C–W systems available in the literature.
3.4.1. B–W Systems
In [33], the suitability of using W-matrix coating materials supersaturated with B was applied to stainless steel substrates. All the W–B coated (including W–13%B and W–23%B) materials are found to be nearly an order of magnitude more resistant to material loss through corrosion–wear compared to uncoated substrates. Wear resistance and hardness of W sequentially implanted with 60 keV ions of B (and C as well) at 300–350 K were studied in [34]. For testing the hardness of samples modified by fluences of 1 × 1015–3 × 1015 and 1 × 1016–3 × 1017 ion/cm2 nano- and micro-indentations were used. And composite materials with 1.3–4.5- and 2.0–6.7-times improved hardness and wear resistance, respectively, were obtained.
Coatings containing high-temperature tungsten boride phases W2B and β-WB were obtained [35] on titanium and steel targets by a shaped charge explosion. In some regions of the target, microhardness reaches 42 GPa.
The stability and hardness of the highest boridesof W, which are built of borophenes separated by metal atoms, were explored in [36].
3.4.2. C–W Systems
Possibilities for the ultrafast compacting of binderless tungsten carbide by combining limited AC and uniaxial pressure-assisted sintering processes were explored in [37].
As is known, the calculation of entropy, a key thermodynamic characteristic, allows the study of system equilibrium, in particular contact interaction during composite material impregnation. The paper [38] introduces the method for calculating the entropy of the tungsten carbide phases WC and W2C in their eutectic alloy by means of scanning their digitized photo-images and further statistical processing. The obtained values of the entropy of phases of WC and W2C, respectively, 29.22 and 58.38 J/mol∙K, agree satisfactorily with the reference data of 32.21 and 56.28 J/mol∙K [39].
3.4.3. B–C–W Systems
TiB2–WC ceramic-metal composite tool materials were fabricated [40] using Co, Ni, and (Ni,Mo) alloy as sintering additives by vacuum hot-pressing technique.
Microstructure, mechanical and tribological properties of WC–TiB2 composites sliding against stainless steel were investigated in [41]. The specific wear rate of WC–TiB2 composites increased with increasing normal load and sliding velocity. WC–30 wt.%TiB2 composite showed better tribological properties than WC–10 wt.%TiB2 and WC–20 wt.%TiB2 composites.
3.4.4. B4C–W Systems
B4C–W composites with 5, 10, 15, and 20 vol.% metallic tungsten were produced [42] by the SPS technique at temperatures of 1500–1600 °C with a 100 °C/min heating rate from room temperature under vacuum by applying 40 MPa uniaxial pressure for 4 min. Commercial B4C and W powders with average particle sizes of 0.7 and 60 μm and of 99.5 and 99.8% purities, respectively, were used. The powder mixture was prepared by milling with WC balls in ethanol medium for 24 h. The formation of the W2B5 phase as a result of the reaction between B4C and W particles was observed. With higher W addition, graphite phase also occurs. An increase in the graphitic C quantity deteriorates mechanical properties. Adding metallic W improves the sinterability and densification of the B4C matrix. Gamma- and neutron-attenuation properties of boron carbide-tungsten B4C–W composite samples with almost theoretical densities were investigated. It is concluded that increasing W share in the B4C–W system causes higher gamma-attenuation behavior and provides lower thermal neutron shielding capacity compared to monolithic B4C.
For B4C ceramics, pressureless sintering is the method with the most industrialized application value. However, it is impossible to sinter them to high densities without special additives promoting densification. Such sintering additives, including metallic W, were described in the review [43].
Sintered composites of WC–WB–W2B were prepared [44] from B4C–W–WC powders using a reactive energization hot-pressing technique that initiated a solid-state reaction between B4C and W. Below a WC mole fraction of 0.769 in the starting powder, WB and WC phases were formed according to the reaction B4C + 5W + xWC = WB + (1 + x)WC, while WB, W2B, and WC phases were produced above a mole fraction of 0.854. Densely sintered bodies were not obtained for pure WC. WC–WB–W2B compositions obtained at WC mole fractions between 0.854 and 0.956 were fully consolidated, having high Young’s modulus and Vickers hardness values and a fracture toughness comparable to those of WC.
Fine-grained WC–Co hard metal grades with additives such as boron carbide B4C were prepared [45] in a sinter furnace. The results of their investigation showed that the grade doped with B4C as a growth inhibitor exhibits more hardness than other doped alloys.
The method of synthesizing B4C coatings by using an RF (Radio Frequency) plasma source with an external magnetic field was described in [46]. The nanohardness of coated steel surfaces was in the range of 14.0–16.6 GPa. It was found that the B4C coatings have 1.73–3.89 times higher hardness than uncoated bare steels serving as targets. This technique seems to be useful for coating W substrates with B4C.
3.4.5. B4C–WB2 Systems
Dense B4C–WB2 composites were fabricated at 1950 °C using B4C and WB2 as raw materials via a hot pressing method [47]. The obtained B4C–68.7 vol.%WB2 composite demonstrated good comprehensive properties with a high flexural strength of 696 MPa, superior hardness of 34.8 GPa, and acceptable fracture toughness of 3.3 MPa∙m1/2. In addition, B4C–WB2 composites demonstrated a good electrical conductivity of 3.3 × 105 S/m, which, together with a low density of 5.589 g/cm3, makes them of interest for cutting tools and armor protection applications.
Dense B4C–TiB2 ceramics were typified [48] by the development of a core/shell structure of the boride grains, with the shell comprising a (Ti,W)B2 solid solution with different assemblages and a variable amount of W guest cation depending on the processing route. The revealed chemical and morphological differences were associated with the presumed densification mechanisms.
3.4.6. B4C–W2B5 Systems
The investigation [49] was undertaken to study the effect of WC and TiC additions to B4C–B–Si mixtures on the sintering behavior and mechanical properties. Powders milled for 2 and 4 h contained 39 and 47 wt.% of WC and TiC, respectively. During heating at temperatures of 800–1500 °C of milled components, they reacted to form B4C, TiB2, and W2B5. In particular, the formation of the W2B5 phase starts at 1100 °C. The maximum amount of W2B5 was reached at temperatures between 1600 and 1800 °C. At higher temperatures, the amount of W2B5 decreases strongly. TiB2 was formed at 1050 °C. At temperatures above 1800 °C, a strong increase in the amount of TiB2 was observed. This increased amount of TiB2 and decreased amount of W2B5 at the same temperature. Three different phases can be observed: B4C, W2B5, and TiB2 (tungsten and titanium boride grain sizes are about 1 μm). Alloy prepared from powder attrition milled for 2 h contained 72, 8, and 20 vol.% of these phases, respectively. The porosity was less than 3 vol.%. The decrease in density with increasing temperature is caused by the dissolution of W2B5 and the formation of (Ti,W)B2. This reaction lowered the hardness as well as the bend strength.
B4C and composite B4C–W2B5 ceramics were prepared [50] via hot pressing. Results show that the values of electrical conductivity for the B4C–W2B5 composite ceramic are much higher than those for the B4C ceramic, but the Seebeck coefficient values are somewhat lower. Both electrical conductivity and the Seebeck coefficient increase with increasing temperature. The thermoelectric figure-of-merit of the composite is higher than that of B4C, though the thermal conductivity of B4C ceramic is somewhat lower than that of the composite one.
An experimental study on the preparation of two tungsten borides, WB and W2B5, was conducted [51] by SHS (Self-propagating High-temperature Synthesis), during which borothermic reduction of WO3 and interaction of W with boron proceeded concurrently. Powder mixtures with two series of molar proportions of WO3:B:W = 1:5.5:x (with x = 1.16–2.5) and 1:7.5:y (with y = 0.5–1.33) were adopted to fabricate WB and W2B5, respectively. The starting stoichiometry of the reactant compact substantially affected the combustion behavior and the phase composition of the final product. The increase in metallic tungsten and boron reduced the overall reaction exothermicity, leading to a decrease in both combustion temperature and reaction front velocity. The initial composition of the reactant compact was optimized for the synthesis of WB and W2B5. In addition to small amounts of W2B and W2B5, the powder compact of WO3 + 5.5B + 2W produced WB dominantly. Optimum formation of W2B5 was observed in the sample of WO3 + 7.5B + 0.85W. Experimental evidence indicates that an excess amount of boron, about 10–13%, is favorable for the formation of WB and W2B5.
Boron carbide-based ceramics were pressureless sintered [52] to a relative density of 96.1% at 2150 °C, with the co-incorporation of tungsten carbide WC and pyrolytic carbon C. The as-batched boron carbide powder was 7.89 m2/g in surface area. A level of fracture toughness as high as 5.80 ± 0.12 MPa∙m1/2 was achieved in the BW–6C composite. Sintering aids of carbon and tungsten boride were formed by an in situ reaction. The toughness improvement was attributed to the presence of thermal residual stress as well as the W2B5 platelets. This study demonstrated that B4C–W2B5 composites could be potential candidate materials for structural applications.
In-situ-formed W2B5 and graphite containing B4C-composites were produced [53] by the SPS technique. A total of 5 vol.% W containing B4C starting powders were shaped into bulk samples at temperatures of 1500, 1550, and 1600 °C under vacuum. Composites with high hardness and improved fracture toughness were obtained.
B4C–(W,Ti)C ceramic composites with different contents of solid-solution (W,Ti)C were produced [54] by hot pressing. Results showed that a chemical reaction took place for this system during hot pressing and resulted in a B4C–TiB2–W2B5 composite with high density and improved mechanical properties compared to monolithic B4C ceramic. Densification rates of the B4C-based ceramic composites were found to be affected by the addition of (W,Ti)C. Increasing (W,Ti)C content led to an increase in the densification rates of the composites. The sintering temperature was lowered from 2150 °C for monolithic B4C to 1850 °C for the B4C–(W,Ti)C composites. The fracture toughness and flexural strength continuously increased with increasing (W,Ti)C content up to 50 wt.%, while the hardness decreased with increasing (W,Ti)C content.
Many of the available information about obtaining methods, structures, and physical and chemical properties of tungsten carbides and their alloys with other materials, including boron carbides, is collected in the monographic handbook [55].
4. Hexagonal Boron Nitride–Iron and Magnetite Composites
4.1. Test Samples Preparation
As it has been mentioned in the Introduction, a number of methods previously developed by us to obtain h-BN nanopowders doped (intercalated or coated) with Fe or Fe3O4 magnetic clusters have been described elsewhere. So, here we present the synthesis procedures only for four series of composite samples that will be characterized below by their chemical and phase compositions, component crystalline structure, and morphology.
Method 1—obtaining the h-BN–Fe composite by iron reduction. Approximately 100 mL of a 50% alcohol solution, 5 g of h-BN powder, and 5 g of FeSO4∙7H2O are placed in a three-necked 250 mL flask equipped with a thermometer, gas tube, and dropping funnel. The reaction mixture is cooled with ice water. Argon is pumped into the flask, and after 30 min of stirring, a 0.5 M NaBH4 solution in ethanol is added drop-wise. Molar ratio: Fe+2:NaBH4 = 1:10. The reduction time is about 120 min. The obtained black precipitate is attached to the bottom of the flask with a magnet and washed three times with ethanol. Then, the suspension is filtered under an argon stream and dried in vacuum (2–4 mmHg) at 70 °C for 4 h.
Method 2—obtaining the h-BN–Fe composite by iron pentacarbonyl decomposition. Note that all these operations are carried out in a fume cupboard in full compliance with safety rules because iron carbonyl is a strong poisonous substance. Approximately 3 g of h-BN is placed in a teflon test tube, and 3 mL of iron(O) pentacarbonyl is added. The test tube is placed in a 0.5 L iron reactor in the teflon-lined autoclave. The autoclave is heated at a rate of 50 °C/min and kept at 230 °C for 2 h. After that, the autoclave is connected to the vacuum system and vacuumed for 2 h at 120–140 °C (2–4 mmHg). The composite powder is washed three times with ethyl alcohol and dried in a vacuum (2–4 mmHg) at 70 °C for 4 h. A black magnetic powder is obtained, which is stored in a desiccator.
Method 3—obtaining of h-BN–Fe3O4 composite by co-precipitation of iron(II) and iron(III) compounds. Approximately 2.8 g of h-BN, 0.7 g of FeSO4∙7H2O, and 1.3 g of FeCl3∙6H2O are placed in a 250 mL three-necked flask equipped with a gas tube, thermometer, and dropping funnel. Argon is pumped into the flask, and 100 mL of freshly distilled water is added. The suspension is heated to 70 °C and after 30 min, 15 mL of a 25% ammonium hydroxide (NH4OH) solution is added drop-wise. The resulting black suspension is stirred for 1 h at 75 °C. The reaction mixture is cooled to room temperature. The obtained black precipitate is attached to the bottom of the flask with a magnet and washed three times with water and ethanol. Then, it is filtered in an argon stream, and theprecipitate is washed again with anhydrous ethanol. The wet mass is dried in vacuum at 120 °C (6 h and 2–4 mmHg). In this way, the brownish-black powder of h-BN–Fe3O4 is obtained. Itis kept in a desiccator.
Method 4—obtaining the h-BN–Fe3O4 composite by iron pentacarbonyl decomposition. Approximately 3 g of h-BN is placed in a teflon flask, and 2.5 mL of iron(O) pentacarbonyl is added. The mixture is stirred on a magnetic stirrer for 2 h at room temperature in an argon atmosphere. A total of 2 mL of water is added to the reaction mixture and placed in a 0.5 l high-pressure argon-pressure reactor, the autoclave, the inner surface of which is covered with teflon. Typical operational conditions are a temperature of 200 °C and a holding time of 2 h. After that, the autoclave is connected to the vacuum system and vacuumed for 2 h at 120–140 °C. A black magnetic powder is obtained, which is stored in the desiccator.
FTIR spectra are tested on five series of samples (their magnetization curves were obtained previously; see below):
Sample 1 is h-BN–Fe composite obtained by the reduction of FeSO4 through sodium borohydride in an argon atmosphere.
Sample 2 again is h-BN–Fe composite, but synthesized by reduction of precursor h-BN–Fe2O3 composite with hydrogen according to the following scheme of reactions: Fe+3 → Fe(OH)3 → FeOOH → Fe2O3 → Fe.
Sample 3 is h-BN–Fe3O4 composite obtained by co-precipitation of iron(II) and iron(III) compounds in the presence of h-BN. This method is based on the chemical reaction: FeSO4 + 2FeCl3 + 8NH4OH → Fe3O4 + (NH4)2SO4 + 6NH4Cl + 4H2O.
Sample 4 is iron-deposited hexagonal boron nitride h-BN–Fe produced from iron(O) pentacarbonyl in the autoclave.
Sample 5 is magnetite-deposited hexagonal boron nitride h-BN–Fe3O4 similarly produced from iron(O) pentacarbonyl in the autoclave, but in the presence of water.
4.2. Pure Hexagonal Boron Nitride
Precursor hexagonal boron nitride powder is chemically synthesized from the liquid charge of boric acid, polyvinyl alcohol, water, and monoethanolamine.
Figure 7 shows its X-ray diffractogram. As can be seen from the diffractogram, the pure, in the sense of undoped, h-BN serving as precursor material has a hexagonal structure and other structural phases are not detected. According to the Scherer formula, these reflections indicate that the particle (grain) size of this phase is in the range of 90–200 nm.
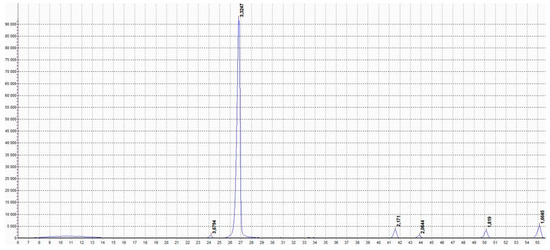
Figure 7.
XRD pattern of pure precursor h-BN. Labels from left to right: 3.6794, 3.3247, 2.1710, 2.0644, 1.8190, and 1.6645.
In general, the exfoliation of 2D materials like h-BN (such as graphite, several-layer graphene and its oxides, molybdenum and tungsten disulfides, metal carbides and carbonitrides, and Mxenes) is possible to conduct by a number of methods.
In particular, for h-BN, the simplest method of hexagonal layer separation and activation is to heat the material to temperatures up to 900–1000 °C in the air. As a result of its partial oxidation, the B–OH functional groups are formed, which help to increase the interlayer distance and, in this way, facilitate the intercalation of foreign, in our case, Fe and Fe3O4 phases.
The process is carried out as follows: 15 g of h-BN is placed in a corundum jar heated in air for 2 h. The powder sample mass increase due to oxidation is up to ~20%. After cooling, the activated h-BN is milled to a nanosize, and the obtained nanopowder is boiled in ethyl alcohol to remove the boron oxide formed during the high-temperature treatment.
To avoid boiled nanoparticle agglomeration, the material should be pre-grinded in an ultrasonic homogenizer before starting its intercalation with iron or its compounds.
Figure 8 shows the diffractogram of the product obtained by pure h-BN thermal decomposition and activation.
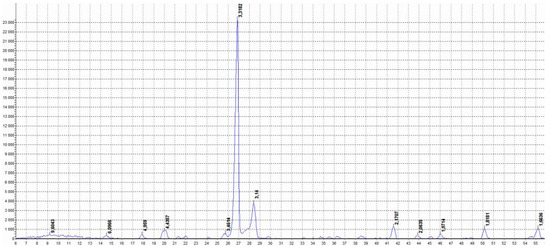
Figure 8.
XRD pattern of thermally exfoliatedand activated h-BN. Labels from left to right: 9.6043, 6.0966, 4.9590, 4.4357, 3.4624, 3.3182, 3.1400, 2.1707, 2.0639, 1.9714, 1.8181, and 1.6636.
It can be seen that during the thermal treatment, the main phase, i.e., h-BN, maintains its structure, although the appearance of some other phases also takes place. They could be caused by the formation of boron oxide and certain functionalized structures during the thermal treatment process.
4.3. Hexagonal Boron Nitride–Iron Composite
In the process of reducing iron sulfate (FeSO4) with sodium borohydride (NaBH4) in the presence of hexagonal boron nitride (h-BN) in an argon atmosphere, all three phases are directly obtained.
The product composition depends on the FeII:NaBH4 molar ratio and air exposure time. The h-BN–Fe composite is formed by rapid filtration and vacuum drying of the reaction mixture. XRD pattern of the obtained composite (Figure 9) reveals reflexes characteristic of h-BN. But the reflexes of zero-valent nanoiron, like other iron compounds, in particular Fe3O4, are not visible. However, iron presence is evident from EDX analysis (Figure 10), magnetic properties of the material (see below Sample 1), and the fact that its interaction with hydrochloric acid (HCl) water solution releases hydrogen gas.

Figure 9.
XRD pattern of h-BN–Fe composite obtained by reduction of FeSO4 with NaBH4 in Ar atmosphere.
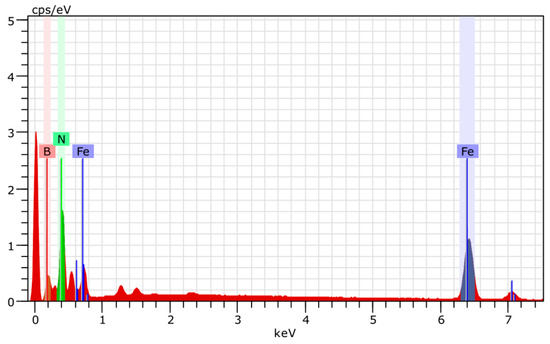
Figure 10.
EDX analysis of h-BN–Fe composite obtained by reduction of FeSO4 with NaBH4 in Ar atmosphere.
From the EDX mapping of chemical element distributions (Figure 11) in the h-BN–Fe composite obtained by the reduction of FeSO4 with NaBH4 in the Ar atmosphere, one can see a quite uniform distribution of B, N, and Fe atoms.

Figure 11.
EDX mapping of all elements (a) and separately B (b), N (c) and Fe (d) distributions in h-BN–Fe composite obtained by reduction of FeSO4 with NaBH4 in Ar atmosphere.
Figure 12 represents a SEM image of the h-BN–Fe composite obtained by thermal decomposition of iron pentacarbonyl Fe(CO)5 in presence of h-BN.

Figure 12.
SEM image of h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in presence of h-BN.
Here we do not show the corresponding diffractogram because it is almost identical with that for material obtained by the reduction process: there are only h-BN, not Fe, peaks. However, iron presence is evident from EDX analysis (Figure 13), magnetic properties of the material (see below Sample 4), and hydrogen gas release as a result of its interaction with HCl solution.
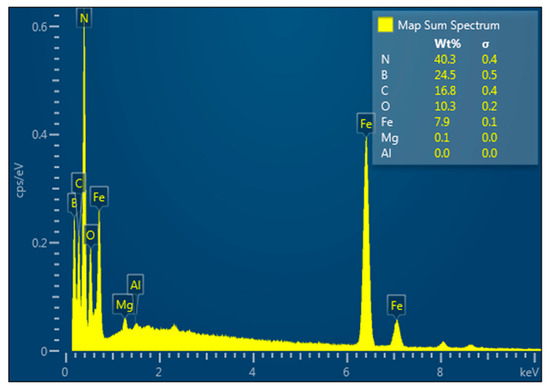
Figure 13.
EDX analysis of the h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in presence of h-BN.
From the EDX mapping of chemical elements (Figure 14) in the h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in the presence of h-BN, one can see a quite uniform distribution of B, N, and Fe atoms, and O as well.

Figure 14.
EDX mapping of all elements (a) and separately N (B is almost identical) (b), Fe (c), and O (d) distributions in h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in presence of h-BN.
Table 1 shows the elemental composition of the h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in the presence of h-BN.

Table 1.
Chemical composition of h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in presence of h-BN (wt.%).
As for Table 2, it reflects the element distribution map sum spectrum in the h-BN–Fe composite obtained by thermal decomposition of Fe(CO) 5 in the presence of h-BN.
Table 2.
Elements distribution map sum spectrum in h-BN–Fe composite obtained by thermal decomposition of Fe(CO)5 in presence of h-BN.
4.4. Hexagonal Boron Nitride–Magnetite Composite
Obtaining the h-BN–Fe3O4 composite by co-precipitation of two- and three-valent iron compounds is based on the chemical reaction: FeSO4 + 2FeCl3 + 8NH4OH → Fe3O4 + (NH4)2SO4 + 6NH4Cl + 4H2O. In its XRD pattern (Figure 15), together with theh-BN strong peak, one can see a low-intensity Fe3O4 peak as well.
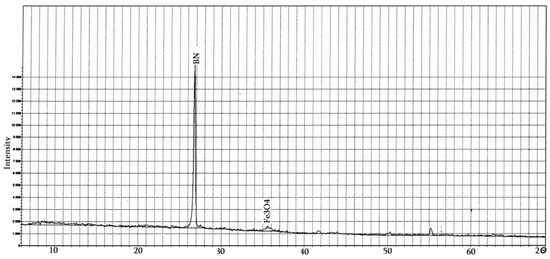
Figure 15.
XRD pattern of the h-BN–Fe3O4 composite obtained by co-precipitation of FeII and FeIII compounds in presence of h-BN.
Figure 16 and Figure 17, respectively, present the SEM image and EDX spectrum of the h-BN–Fe3O4 composite obtained by the co-precipitation method.

Figure 16.
SEM image of h-BN–Fe3O4 composite obtained by co-precipitation of FeII and FeIII compounds in presence of h-BN.
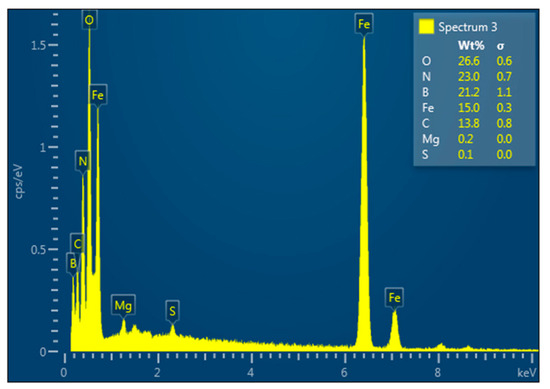
Figure 17.
EDX analysis of the h-BN–Fe3O4 composite obtained by co-precipitation of FeII and FeIII compounds in presence of h-BN.
As for the diffractogram of the h-BN–Fe3O4 composite obtained by thermal decomposition of Fe(CO)5 in the presence of h-BN and water H2O, it is not shown because it is almost identical with that obtained by co-precipitation: there are only h-BN and weak Fe3O4 peaks.
According to the SEM image (Figure 18), the h-BN–Fe3O4 composite obtained by thermal decomposition of Fe(CO)5 in the presence of h-BN and H2O consists of a substance with a mesh structure surrounded by hexagonal boron nitride particles.

Figure 18.
SEM image of the h-BN–Fe3O4 composite obtainedby thermal decomposition of Fe(CO)5 in the presence of h-BN and H2O.
Figure 19 presents the EDX spectrum of the h-BN–Fe3O4 composite obtained by thermal decomposition of Fe(CO)5 in the presence of h-BN and H2O.
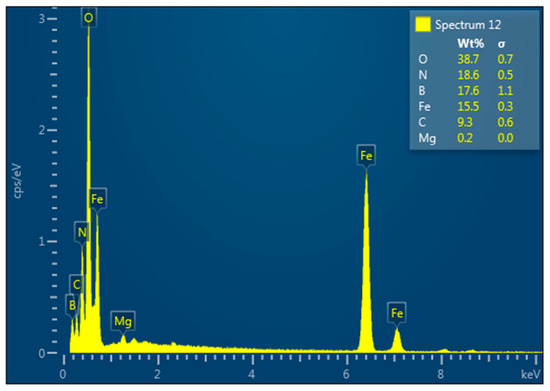
Figure 19.
EDX analysis of the h-BN–Fe3O4 composite obtained by thermal decomposition of Fe(CO)5 in presence of h-BN and H2O.
4.5. Fourier Transform Infrared Spectra
FTIR spectroscopy is used to determine the chemical and structural nature of the obtained h-BN–Fe and h-BN–Fe3O4composite materials. Namely, Figure 20 depicts the room-temperature FTIR spectra of Samples 1, 2, 3, 4, and 5, the synthesizing routes of which are described above.
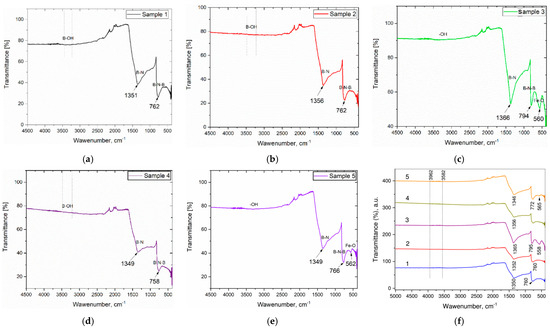
Figure 20.
FTIR spectra of h-BN–Fe and h-BN–Fe3O4 composite samples.
In the FTIR spectrum of Sample 1, the h-BN–Fe composite, strong and broad peaks at 1351 and 762 cm−1 are observed, apparently [56,57] due to B–N stretching and out-of-plane B–N–B bending vibrations, respectively. These two peaks are extremely typical of sp2 bonds inside h-BN, and their presence provides essential proof of h-BN production [58].
The placement of two primary boron nitride peaks is determined by the synthesis process and conditions, the crystal structure and layered state of the sample, the physical-chemical properties of the composite mixture, etc. [59]. The broad absorption bands at around 3420–3250 cm−1 have to be related to B–O–H stretching of absorbed water molecules [60].
Spectra of Samples 2 and 4, again the h-BN–Fe composites, also show two main peaks characteristic of hexagonal boron nitride at 1356 and 762 and 1349 and 758 cm−1, which are related to B–N tension and B–N–B bending vibrations, respectively. In Sample 2, the fingerprint peak characteristic of maghemite (Fe2O3) is not clearly observed, indicating the reduction of iron oxide with hydrogen. It is known that such reduction depends on various factors, including the size of nanoparticles, reaction routes, processing conditions, etc. [61]. The broad absorption bands at around 3400–3200 cm−1 in both samples could be ascribed to the hydroxyl group (–OH) vibrations.
In the FTIR spectra of Samples 3 and 5, the h-BN–Fe3O4 composites exhibit characteristic peaks for hexagonal boron nitride, like the samples reported above: 1366 and 794, and 1349 and 766 cm−1, respectively. In comparison to Samples 1 and 2, the FTIR spectra of these samples reveal extra peaks at 560 and 562 cm−1, which are characteristic of the Fe–O bond [60], indicating that these materials are composed of h-BN and Fe3O4. Note that the exact positions and intensities of the peaks are a combination of both h-BN and magnetite and can vary depending on the concentration of magnetite dopants and their specific crystallographic sites within the h-BN lattice [60,62]. Another absorption peak at around 3200 cm−1 could again be ascribed to the hydroxyl group (–OH) vibrations.
4.6. Magnetic Properties
Previous work [63] aimed to investigate the magnetization of obtained h-BN nanopowders doped with magnetic nanoclusters in the form of ferromagnetic metallic iron Fe or its ferrimagnetic oxide Fe3O4—magnetite.
As is known, just the magnetization curve, i.e., magnetization M as a function M = M(H) of the applied magnetic field H, provides important information about material magnetic properties, including saturation MS and remnant MR magnetizations, and coercive force HC as well.
When analyzing M = M(H) characteristics, one has to take into account that the magnetic parameters of ferro- and ferrimagnetic powder materials depend on particle size. Above a critical diameter of Dc1, particles of such a magnetic material exhibit multidomain properties, while below this size they pass into a monodomain state, and below a critical diameter of Dc2, they exhibit superparamagnetism [64,65].
Figure 21 shows the room-temperature magnetization curves with hysteresis loops for h-BN–Fe and h-BN–Fe3O4 composite Samples 1, 2, 3, 4, and 5, the synthesizing routes of which are described above.
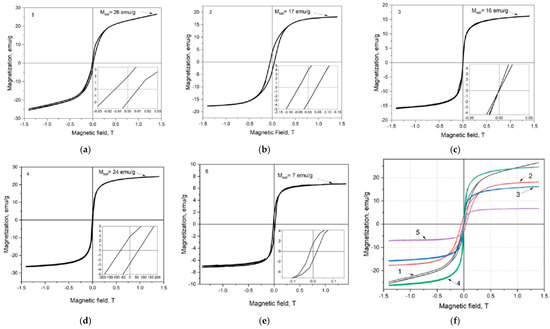
Figure 21.
Magnetization curves of h-BN–Fe and h-BN–Fe3O4 composite samples [63].
Note that the magnetization curves of Samples 1, 2, and 5 exhibit hysteresis loops with similar shapes. Their magnetization increases with the magnetic field, initially rapidly up to an external field of approximately 5 kG and then gradually with a rising field. At the maximum applied magnetic field of 14 kG, the magnetizations of these samples are MHmax = 26.5, 18.0, and 6.75 emu/g, respectively. We should emphasize that the magnetization of Sample 1 is not completely saturated at the maximum value of the applied external magnetic field. As for Sample 2, one can see that its MHmax value is very close to saturation. And for Sample 5, the MHmax value coincides with MS. For these three samples, MR and HC were equal to 1.85, 3.80, and 1.76 emu/g and 114, 456, 164 G, respectively.
As MS and MR values, in general, increase with material crystallinity and the variation of the coercivity can be caused by various combined factors such as atom distribution in the magnetic phase structure and crystallite size and distribution in the composite [64,66], it is workable to notice that these samples exhibit similar magnetic behavior, characteristic of soft magnetic materials. Among them, Sample 1 is closest to the behavior of superparamagnetic material.
The room-temperature magnetization hysteresis loops corresponding to Samples 3 and 4 are S-shaped and extremely thin due to the absence of remnant magnetization and negligible coercivity. More precisely, in Sample 3, there is actually no coercivity (MHmax = 16 emu/g), and, therefore, this nanocomposite exhibits superparamagnetic behavior. As for Sample 4, there is a small coercivity (HC = 63 G and MHmax = 24.5 emu/g), and in behavior, it shows similarities with Sample 1.
Summarizing magnetic property measurement for h-BN–Fe and h-BN–Fe3O4 composites, let us state that iron valent states—FeO, FeIIFe2IIIO4, and Fe2IIIO3—characteristics of free metallic iron Fe, magnetite Fe3O4, and maghemite Fe2O3, respectively, lead to ferro-, ferri-, and antiferromagnetic properties. Consequently, hexagonal boron nitride h-BN doped with Fe has to be the best magnetic among the studied three composites. But using h-BN–Fe in BNCT is problematic because of the fast oxidation of iron in aqueous media. As for h-BN–Fe2O3, it is not a magnetic material. Thus, the magnetite-doped hexagonal boron nitride h-BN–Fe3O4 could be considered an optimal choice.
In this regard, we mention the effect of fast (with an energy of 167 MeV) heavy (132Xe) ion streams of intensities of 4.14 × 1012, 2.75 × 1013, and 3.83 × 1014 ion/cm2 on the 20–30 nm Fe3O4 magnetic particles. Recently, it was found [67] that when irradiated in this way, nanoparticles change their magnetic properties depending on the irradiation intensity. This fact could be taken into account if not only BNCT agent delivery but also its extraction are intended to be controlled by the external magnetic field.
According to the preliminary studies [68], 250 keV proton beams of intensity ~1016 p/cm2 do not seriously affect our sample structure. And as proton and neutron have almost equal masses, this means that irradiation with (epi)thermal (of energy less than ~1 keV) neutrons would not lead to their ballistic degradations; all the neutron-radiation structural defects could be related to the 10B–n nuclear reactions. Consequently, neutron irradiation during a BNCT clinical procedure would not affect our material’s magnetic properties, unlike the above-described situation, when Fe3O4 nanoparticles are influenced by a heavy-ion high-energy stream.
5. Conclusions
In summary, there are developed novel obtaining methods for two boron-containing composite materials intensively interacting with neutron radiation: boron carbide–tungsten B4C–W sandwich structures and hexagonal boron nitride nanopowders doped with magnetic clusters of iron or magnetite h-BN–(Fe,Fe3O4).
Boron carbide–tungsten B4C–W composites are obtained by three different methods. First, tungsten W film is deposited on the boron carbide B4C surface by its coating from a preliminary synthesized peroxotungstic acid solution carried out by the dipping technique and further multistage thermal treatment to reduce tungsten. Second, tungsten W powder is compacted with a boron carbide B4C crystal surface. And third, boron carbide B4C powder is compacted with tungsten W foil.
Hexagonal boron nitride nanopowders doped with iron or magnetite clusters h-BN–(Fe,Fe3O4) are obtained by four different methods. First, metallic iron Fe is reduced from iron sulfate FeIISO4 in the presence of hexagonal boron nitride h-BN powder. Second, two-valent iron FeII and three-valent iron FeIII compounds, iron sulfide FeIISO4 and iron chloride FeIIICl3, are co-precipitated in the presence of hexagonal boron nitride h-BN powder. Third, iron pentacarbonyl FeO(CO)5 is decomposed in the presence of hexagonal boron nitride h-BN powder. And fourth, the same method is used, but in the presence of H2O.
The chemical and phase compositions, component crystalline structure, and composite morphology, as well as some magnetic properties of materials obtained by the proposed methods, have been characterized (using various techniques such as XRD, EDX, SEM, FTIR, and VSM). Based on the observed properties, it can be shown that B4C–W and h-BN–(Fe,Fe3O4) composites are suitable for use in neutron shielding and neutron therapy, respectively.
Namely, our boron carbide-tungsten B4C–W composites produced in sandwich/layered form respond to the main challenge of boron-based neutron shielding, such as the gamma-radiation phenomenon accompanying the absorption of neutrons by 10B nuclei. Layers of tungsten, as a heavy metal, effectively attenuate the gamma radiation that accompanies the absorption of neutrons in the boron carbide shielding layers.
As for the obtained hexagonal boron nitride nanopowders doped with nanoclusters of ferromagnetic iron or ferrimagnetic magnetite h-BN–(Fe,Fe3O4), they respond to the main challenge of boron-neutron capture therapy using boron-rich nanosystems for delivery agents of 10B nuclei: how to provide their preferential storage in cancer cells. The presence of magnetic clusters in these therapeutic agents allows their targeted delivery to tumor tissues using an external magnetic field, thus avoiding damage to healthy tissues due to neutron absorption.
Thus, several new methods have been developed for the synthesis of boron compound-matrix composites; some of them have actually been obtained for the first time, and their properties have been studied, showing that composites with boron-containing matrices, which intensively absorb neutrons, could find application in neutron-shielding and neutron-therapy techniques.
Author Contributions
Conceptualization, L.C. and R.C.; methodology, L.C., O.T., M.M., S.K. and R.C.; software, S.M., N.G., T.B., V.M. and J.M.; validation, T.M. and K.D.; formal analysis, T.D., K.K. and R.T.; investigation, L.C., S.M., N.G., O.T., M.M., V.M., J.M. and N.B.; resources, L.C., O.T., M.M., S.K. and R.C.; data curation, T.B., J.M., T.M., K.D., T.D., K.K., R.T. and R.C.; writing—original draft preparation, L.C., N.B. and R.C.; writing—review and editing, L.C.; visualization, O.T., T.B., V.M. and J.M.; supervision, L.C.; project administration, L.C.; funding acquisition, S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Shota Rustaveli National Science Foundation of Georgia, grant number PHDF-22-1299, and the APC was funded by PHDF-22-1299.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chkhartishvili, L.; Chedia, R.; Tsagareishvili, O.; Mirzayev, M.; Makatsaria, S.; Gogolidze, N.; Barbakadze, N.; Buzariashvili, M.; Lekashvili, O.; Jinikashvili, I. Preparation of neutron-capturing boron-containing nanosystems. In Proceedings of the 9th International Conference and Exhibition on Advanced and Nano Materials, Victoria, BC, Canada, 24–26 October 2022; IAEMM: Victoria, BC, Canada, 2022; pp. 1–15, ISBN 978-1-77835-171-6. Available online: https://iaemm.com/Pubdetails.php (accessed on 1 August 2023).
- Chkhartishvili, L.; Makatsaria, S.; Gogolidze, N. Boron-containing fine-dispersive composites for neutron-therapy and neutron-shielding. In Proceedings of the International Scientific-Practical Conference “Innovations and Modern Challenges—2022”, Tbilisi, Georgia, 18–19 November 2022; Publishing House “Technical University”: Tbilisi, Georgia, 2023; pp. 221–226, ISBN 978-9941-28-944-6. Available online: https://publishhouse.gtu.ge/en/ (accessed on 1 August 2023).
- Nabakhtiani, G.; Chkhartishvili, L.; Gigineishvili, A.; Tsagareishvili, O.; Gabunia, D.; Rostomashvili, Z.; Dekanosidze, S. Attenuation of gamma-radiation concomitant neutron-absorption in boron–tungsten composite shields. Nano Stud. 2013, 8, 259–266. Available online: https://www.nanostudies.org/index.php/nano/issue/archive (accessed on 1 August 2023).
- Evans, B.R.; Lian, J.; Ji, W. Evaluation of shielding performance for newly developed composite materials. Ann. Nucl. Energy 2018, 116, 1–9. [Google Scholar] [CrossRef]
- Chkhartishvili, L. Chapter 11. Boron-Containing Nanostructured Materials for Neutron-Shields. In Nanostructured Materials for the Detection of CBRN; Bonca, J., Kruchinin, S., Eds.; Springer Science: Dordrecht, The Netherlands, 2018; pp. 133–154. [Google Scholar] [CrossRef]
- Martin, P.M. Active thin films: Applications for graphene and related materials. Vac. Technol. Coat. 2018, 19, 6–14. Available online: https://digital.vtcmag.com/12727/10466/index.html?20400 (accessed on 1 August 2023).
- Zinovev, A.; Terentyev, D.; Chang, C.-C.; Yin, C.; Bakaev, A.; Rieth, M.; Lied, P.; Reiser, J.; Bonnekoh, C. Effect of neutron irradiation on ductility of tungsten foils developed for tungsten-copper laminates. Nucl. Mater. Energy 2022, 30, 101133. [Google Scholar] [CrossRef]
- Sorokin, O.; Kuznetsov, B.; Lunegova, Y.; Erasov, V. High-temperature composites with a multi-layered structure (Review). Proc. All-Russ. Sci. Res. Inst. Aviat. Mater. 2020, 88, 42–53. (In Russian) [Google Scholar] [CrossRef]
- Mann, K.S.; Mann, S.S. Py-MLBUF: Development of an online-platform for gamma-ray shielding calculations and investigations. Ann. Nucl. Energy 2021, 150, 107845. [Google Scholar] [CrossRef]
- Dai, M.; Zhang, Z.; Zhu, J.; Wang, X.; Xu, J.; Fu, X.; Bai, L.; Huang, Q.; Wang, Z.; Chen, L. Influence of interface roughness on reflectivity of tungsten/boron-carbide multilayers with variable bi-layer number by X-ray reflection and diffuse scattering. Chin. Opt. Lett. 2009, 7, 738–740. Available online: https://opg.optica.org/col/abstract.cfm?uri=col-7-8-738 (accessed on 1 August 2023).
- Makatsaria, S.; Chkhartishvili, L.; Dekanosidze, S.; Chedia, R. Nanopowder boron compounds doped with ferromagnetic clusters for BNCT. Int. J. Adv. Nano Comput. Anal. 2023, 2, 1–12. Available online: https://www.researchlakejournals.com/index.php/IJANCA/article/view/189 (accessed on 1 August 2023).
- Seneviratne, D.S.; Saifi, O.; Mackeyev, Y.; Malouff, T.; Krishnan, S. Next-generation boron drugs and rational translational studies driving the revival of BNCT. Cells 2023, 12, 1398. [Google Scholar] [CrossRef]
- Wang, Y.; Long, B.F.; Liu, C.Y.; Lin, G.A. Evolution of reduction process from tungsten oxide to ultrafine tungsten powder via hydrogen. High Temp. Mater. Process. 2021, 40, 171–177. [Google Scholar] [CrossRef]
- Barbakadze, N.; Sarajishvili, K.; Chedia, R.; Chkhartishvili, L.; Tsagareishvili, O.; Mikeladze, A.; Darchiashvili, M.; Ugrekhelidze, V. Obtaining ultrafine powders of some boron carbide-based nanocomposites using liquid precursors. Nanotechnol. Percept. 2019, 15, 243–256. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Chedia, R.; Tsagareishvili, O.; Barbakadze, N.; Sarajishvili, K.; Darchiashvili, M.; Ugrekhelidze, V.; Korkia, T. Synthesizing fine-grained powders of complex compositions B4C–TiB2–WC–Co. Solid State Sci. 2020, 108, 106439. [Google Scholar] [CrossRef]
- Barbakadze, N.; Chkhartishvili, L.; Mikeladze, A.; Tsagareishvili, O.; Sarajishvili, K.; Korkia, T.; Darchiashvili, M.; Rurua, L.; Jalabadze, N.; Chedia, R. Method of obtaining multicomponent fine-grained powders for boron carbide matrix ceramics production. Mater. Today Proc. 2022, 51, 1863–1871. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Jalabadze, N.; Nadaraia, L.; Korkia, T.; Chedia, R. New low-temperature method of synthesis of boron carbide matrix ceramics ultra-dispersive powders and their spark plasma sintering. Solid State Phenom. 2022, 331, 173–184. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Chedia, R.; Tsagareishvili, O.; Bugdayci, M.; Karagoz, I.; Maras, T.; Jalabadze, N.; Kvatchadze, V. Chapter 4: Combustion synthesis of boron carbide matrix for superhard nanocomposites production. In Advances in Combustion Synthesis and Technology; Bugdayci, M., Oncel, L., Eds.; Bentham Science Publisher: Singapore, 2022; pp. 66–95. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Tsagareishvili, O.; Kvatchadze, V.; Tavkhelidze, V.; Mestvirishvili, Z.; Driaev, D.; Barbakadze, N.; Nadaraia, L.; Sarajishvili, K.; et al. Advanced boron carbide matrix nanocomposites obtained from liquid-charge: Focused review. Condens. Matter 2023, 8, 37. [Google Scholar] [CrossRef]
- Hung, C.-C.; Hurst, J.; Santiago, D.; Rogers, R.B. Exfoliation of Hexagonal Boron Nitride via Ferric Chloride Intercalation (NASA/TM–2014-218125); NASA Glenn Research Center: Cleveland, OH, USA, 2014; pp. 1–20. Available online: https://ntrs.nasa.gov/api/citations/20140005373/downloads/20140005373.pdf (accessed on 1 August 2023).
- Píš, I.; Nappini, S.; Bondino, F.; Menteş, T.O.; Sala, A.; Locatelli, A.; Magnano, E. Fe intercalation under graphene and hexagonal boron nitride in-plane heterostructure on Pt(111). Carbon 2018, 134, 274–282. [Google Scholar] [CrossRef]
- Patel, R.B.; Liu, J.; Eng, J.; Iqbal, Z. One-step CVD synthesis of a boron nitride nanotube–iron composite. J. Mater. Res. 2011, 26, 1332–1339. [Google Scholar] [CrossRef]
- Chang, M.; Leung, C.; Wang, D.N.; Cheng, D. Process for CVD Deposition of Tungsten Layer on Semiconductor Wafer. US Patent 5028565, 2 July 1991. Available online: https://patentimages.storage.googleapis.com/f2/06/0f/1d36151bc50532/US5028565.pdf (accessed on 1 August 2023).
- Kim, S.H. Deposition of tungsten thin film on silicon surface by low pressure chemical vapor deposition method. J. Korean Chem. Soc. 1994, 38, 473–479. [Google Scholar]
- Plyushcheva, S.V.; Mikhailov, G.M.; Shabel’nikov, L.G.; Shapoval, S.Y. Tungsten thin-film deposition on a silicon wafer: The formation of silicides at W–Si interface. Inorg. Mater. 2009, 45, 140–144. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.-H.; Sohn, I.-H.; Hwang, T.-J.; Lee, K.-Y. Preparation of tungsten metal film by spin coating method. Korea–Aust. Rheol. J. 2002, 14, 71–76. Available online: https://www.cheric.org/PDF/KARJ/KR14/KR14-2-0071.pdf (accessed on 1 August 2023).
- Singla, G.; Singh, K.; Pandey, O. Structural and thermal properties of in-situ reduced WO3 to W powder. Powder Technol. 2013, 237, 9–13. [Google Scholar] [CrossRef]
- Yu, M.L.; Ahn, K.Y.; Joshi, R.V. Surface reactions in the chemical vapor deposition of tungsten using WF6 and SiH4 on Al, PtSi, and TiN. J. Appl. Phys. 1990, 67, 1055–1061. [Google Scholar] [CrossRef]
- Gao, J.; Chan, L.H.; Wongsenakhum, P. Methods for Improving Uniformity and Resistivity of Thin Tungsten Films. US Patent 7655567B1, 2 February 2010. Available online: https://patentimages.storage.googleapis.com/0d/c7/0b/6be43db1030817/US7655567.pdf (accessed on 1 August 2023).
- Yang, M.; Aarnink, A.A.I.; Kovalgin, A.Y.; Gravesteijn, D.J.; Wolters, R.A.M.; Schmitz, J. Comparison of tungsten films grown by CVD and hot-wire assisted atomic layer deposition in a cold-wall reactor. J. Vac. Sci. Technol. A 2016, 34, 01A129. [Google Scholar] [CrossRef]
- Dippel, A.-C.; Schneller, T.; Lehmann, W.; Reichenberg, B.; Waser, R. Tungsten coatings by chemical solution deposition for ceramic electrodes in fluorescent tubes. J. Mater. Chem. 2008, 18, 3501–3506. [Google Scholar] [CrossRef]
- Cao, P.; Cao, J.-P.; Cao, J.-H. Boron Carbide Ceramic Metallization Preparation Method. CN Patent CN110981550B, 7 December 2021. (In Chinese). Available online: https://eureka.patsnap.com/pdfnew/?patentId=cede242d-11a6-41e5-880a-e2a2dfb9ae88 (accessed on 1 August 2023).
- Mallia, B.; Dearnley, P. Exploring new W–B coating materials for the aqueous corrosion–wear protection of austenitic stainless steel. Thin Solid Films 2013, 549, 204–215. [Google Scholar] [CrossRef]
- Guldamashvili, A.; Nardaya, Y.; Nebieridze, T.; Sanaia, E.; Sichinava, A.; Kadaria, M. Mechanical properties of tungsten implanted with boron and carbon ions. J. Mater. Sci. Eng. A 2017, 7, 82–88. [Google Scholar] [CrossRef]
- Gromilov, S.A.; Kinelovskii, S.A.; Alekseev, A.V.; Kirienko, I.B. Investigation of W2B and β-WB high-temperature phases in coatings produced by a shaped charge explosion. J. Struct. Chem. 2010, 51, 1126–1131. [Google Scholar] [CrossRef]
- Szwacki, N.G. The structure and hardness of the highest boride of tungsten, a borophene-based compound. Sci. Rep. 2017, 7, 4082. [Google Scholar] [CrossRef]
- Mazo, I.; Molinari, A.; Sglavo, V.M. Electrical resistance flash sintering of tungsten carbide. Mater. Des. 2022, 213, 110330. [Google Scholar] [CrossRef]
- Syrovatko, Y.V. Calculation of the entropy of the eutectic phases WC and W2C in alloy W–C by the method of statistical processing of photo-images. Ukr. Appl. Phys. 2020, 4, 79–84. (In Russian). Available online: https://applphys.orion-ir.ru/appl-20/20-4/PF-20-4-79.pdf (accessed on 1 August 2023).
- Pankratz, L.B. Thermodynamic Properties of Carbides, Nitrides, and Other Selected Substances; U.S. Department of the Interior: Washington, DC, USA, 1994; Bulletin 696, pp. 900–901. Available online: https://digital.library.unt.edu/ark:/67531/metadc12836/ (accessed on 1 August 2023).
- Song, J.; Huang, C.; Zou, B.; Liu, H.; Liu, L.; Wang, J. Effects of sintering additives on microstructure and mechanical properties of TiB2–WC ceramic–metal composite tool materials. Int. J. Refract. Met. Hard Mater. 2012, 30, 91–95. [Google Scholar] [CrossRef]
- Zhang, C.; Song, J.; Jiang, L.; Gao, J.; Liang, G.; Lei, C.; Xie, J.; Wang, S.; Lv, M. Fabrication and tribological properties of WC–TiB2 composite cutting tool materials under dry sliding condition. Tribol. Int. 2017, 109, 97–103. [Google Scholar] [CrossRef]
- Ozer, S.C.; Buyuk, B.; Tugrul, A.B.; Turan, S.; Yucel, O.; Goller, G.; Sahin, F.C. Gamma and neutron shielding behavior of spark plasma sintered boron carbide–tungsten based composites. In TMS 2016 145th Annual Meeting Supplemental Proceedings, Nashville, TN, USA, 14–18 February 2016; The Minerals, Metals & Materials Society, Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 449–456. [Google Scholar] [CrossRef]
- Zhang, W.; Yamashita, S.; Kita, H. Progress in pressureless sintering of boron carbide ceramics—A review. Adv. Appl. Ceram. 2019, 118, 222–239. [Google Scholar] [CrossRef]
- Sugiyama, S.; Taimatsu, H. Preparation of WC–WB–W2B composites from B4C–W–WC powders and their mechanical properties. Mater. Trans. 2002, 43, 1197–1201. [Google Scholar] [CrossRef]
- Tamizifar, H.; Hadian, A.; Tamizifar, M. The Comparison between boron carbide (B4C) with other common inhibitors on physical and mechanical properties of WC/Co. Int. J. Mod. Phys. Conf. Ser. 2012, 5, 102–110. [Google Scholar] [CrossRef]
- Taran, A.V.; Garkusha, I.E.; Taran, V.S.; Muratov, R.M.; Skoblo, T.S.; Sidashenko, O.I.; Romaniuk, S.P.; Maltsev, T.V.; Baturin, A.A. Structure and properties of B4C coatings obtained by RF sputtering with external magnetic field. In Nanomaterials and Nanocomposites, Nanostructure Surfaces, and Their Applications; Fesenko, O., Yatsenko, L., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 51–57. [Google Scholar] [CrossRef]
- Ma, K.; Shi, X.; Cao, X.; Yang, Z.; Zuo, J.; Xu, J.; Li, M. Mechanical, electrical properties and microstructures of hot-pressed B4C–WB2 composites. Ceram. Int. 2022, 48, 20211–20219. [Google Scholar] [CrossRef]
- Silvestroni, L.; Failla, S.; Gilli, N.; Melandri, C.; Savacı, U.; Turan, S.; Sciti, D. Disclosing small scale length properties in core-shell structured B4C–TiB2 composites. Mater. Des. 2021, 197, 109204. [Google Scholar] [CrossRef]
- Hofmann, H.; Petzow, G. Structure and properties of reaction hot-pressed B4C–TiB2–W2B5 materials. J. Less-Common Met. 1986, 117, 121–127. [Google Scholar] [CrossRef]
- Cai, K.-F.; Nan, C.-W. The influence of W2B5 addition on microstructure and thermoelectric properties of B4C ceramic. Ceram. Int. 2000, 26, 523–527. [Google Scholar] [CrossRef]
- Yeh, C.; Wang, H. Preparation of tungsten borides by combustion synthesis involving borothermic reduction of WO3. Ceram. Int. 2011, 37, 2597–2601. [Google Scholar] [CrossRef]
- Yin, J.; Huang, Z.; Liu, X.; Zhang, Z.; Jiang, D. Microstructure, mechanical and thermal properties of in situ toughened boron carbide-based ceramic composites co-doped with tungsten carbide and pyrolytic carbon. J. Eur. Ceram. Soc. 2013, 33, 1647–1654. [Google Scholar] [CrossRef]
- Ozer, S.C.; Turan, S.; Sahin, F.C. Mechanical and microstructural properties of spark plasma sintered B4C−W2B5 composites. In Proceedings of the 18th International Metallurgy and Materials Congress, Istanbul, Turkey, 29 September−1 October 2016; UChTEA: Istanbul, Turkey, 2016; pp. 62–66. Available online: https://www.metalurji.org.tr/index.php/duyurular/529-immc-2016-bildiriler-kitabi-yayinlandi (accessed on 1 August 2023).
- Deng, J.; Zhou, J.; Feng, Y.; Ding, Z. Microstructure and mechanical properties of hot-pressed B4C/(W,Ti)C ceramic composites. Ceram. Int. 2002, 28, 425–430. [Google Scholar] [CrossRef]
- Shabalin, I.L. Ultra-High Temperature Materials IV: Refractory Carbides III (W Carbides). A Comprehensive Guide and Reference Book; Springer Nature: Cham, Switzerland, 2022; pp. i–xiv, 1–934. [Google Scholar] [CrossRef]
- Kostoglou, N.; Polychronopoulou, K.; Rebholz, C. Thermal and chemical stability of hexagonal boron nitride (h-BN) nanoplatelets. Vacuum 2015, 112, 42–45. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Shin, B.-G.; Lee, D.-E.; Yoon, K.-B. Preparation of PP/2D-nanosheet composites using MoS2/MgCl2- and BN/MgCl2-Bi supported Ziegler–Natta catalysts. Catalysts 2020, 10, 596. [Google Scholar] [CrossRef]
- Ortiz, D.G.; Pochat-Bohatier, C.; Cambedouzou, J.; Bechelany, M.; Miele, P. Exfoliation of hexagonal boron nitride (h-BN) in liquid phase by ion intercalation. Nanomaterials 2018, 8, 716. [Google Scholar] [CrossRef]
- Wang, J.; Ma, F.; Liang, W.; Wang, R.; Sun, M. Optical, photonic and optoelectronic properties of graphene, h-BN and their hybrid materials. Nanophotonics 2017, 6, 943–976. [Google Scholar] [CrossRef]
- Dee, G.; O’donoghue, O.; Rafferty, A.; Gannon, L.; McGuinness, C.; Gun’ko, Y.K. Boron nitride nanosheets functionalized with Fe3O4 and CoFe2O4 magnetic nanoparticles for nanofiltration applications. ACS Appl. Nano Mater. 2023, 6, 12526–12536. [Google Scholar] [CrossRef] [PubMed]
- Konopatsky, A.S.; Firestein, K.L.; Evdokimenko, N.D.; Kustov, A.L.; Baidyshev, V.S.; Chepkasov, I.V.; Popov, Z.I.; Matveev, A.T.; Shetinin, I.V.; Leybo, D.V.; et al. Microstructure and catalytic properties of Fe3O4/BN, Fe3O4(Pt)/BN, and FePt/BN heterogeneous nanomaterials in CO2 hydrogenation reaction: Experimental and theoretical insights. J. Catal. 2021, 402, 130–142. [Google Scholar] [CrossRef]
- Bangari, R.S.; Yadav, V.K.; Singh, J.K.; Sinha, N. Fe3O4-functionalized boron nitride nanosheets as novel adsorbents for removal of arsenic(III) from contaminated water. ACS Omega 2020, 5, 10301–10314. [Google Scholar] [CrossRef]
- Makatsaria, S.; Kekutia, S.; Markhulia, J.; Mikelashvili, V.; Chkhartishvili, L.; Chedia, R. Magnetic properties of nanopowder h-BN doped with Fe and Fe3O4 nanoclusters. Nano Stud. 2021–2022, 21/22, 287–292. [Google Scholar] [CrossRef]
- Lee, J.S.; Cha, J.M.; Yoon, H.Y.; Lee, J.-K.; Kim, Y.K. Magnetic multi-granule nanoclusters: A model system that exhibits universal size effect of magnetic coercivity. Sci. Rep. 2015, 5, 12135. [Google Scholar] [CrossRef]
- Binns, C. Chapter 1—Tutorial section on nanomagnetism. Front. Nanosci. 2014, 6, 1–32. [Google Scholar] [CrossRef]
- Avancini, T.G.; Souza, M.T.; de Oliveira, A.P.N.; Arcaro, S.; Alves, A.K. Magnetic properties of magnetite-based nano-glass-ceramics obtained from a Fe-rich scale and borosilicate glass wastes. Ceram. Int. 2019, 45, 4360–4367. [Google Scholar] [CrossRef]
- Darziyeva, T.A.; Alekperov, E.S.; Jabarov, S.H.; Mirzayev, M.N. Influence of heavy ions on the magnetic properties of Fe3O4 nanoparticles. Integr. Ferroelectr. Int. J. 2023, 232, 127–133. [Google Scholar] [CrossRef]
- Mirzayev, M. Personal Communication; Institute of Radiation Problems: Baku, Azerbaijan, 2023. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).