
Ab Initio Study of Structural, Electronic, and Thermal Properties of Pt/Pd-Based Alloys

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
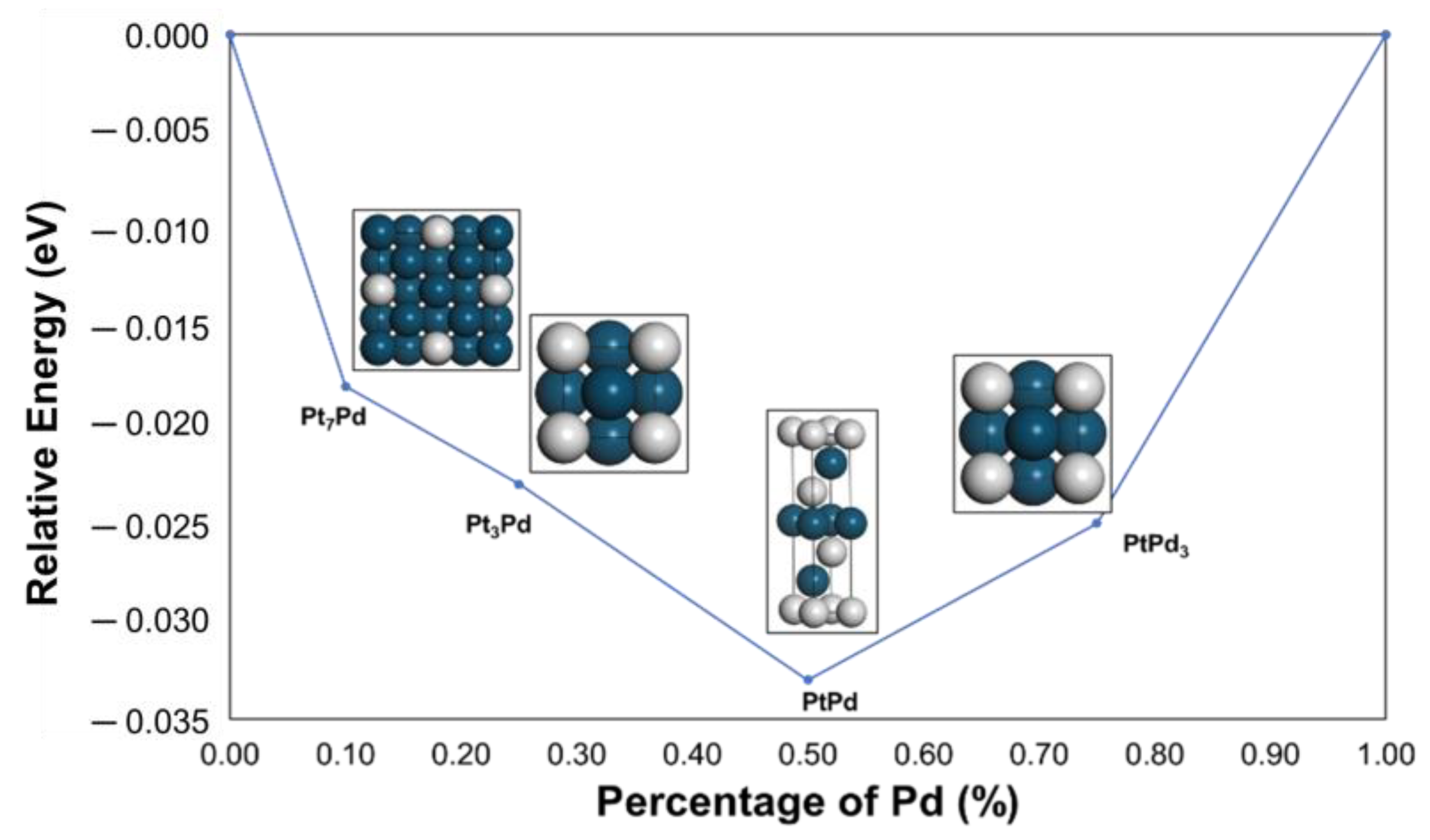
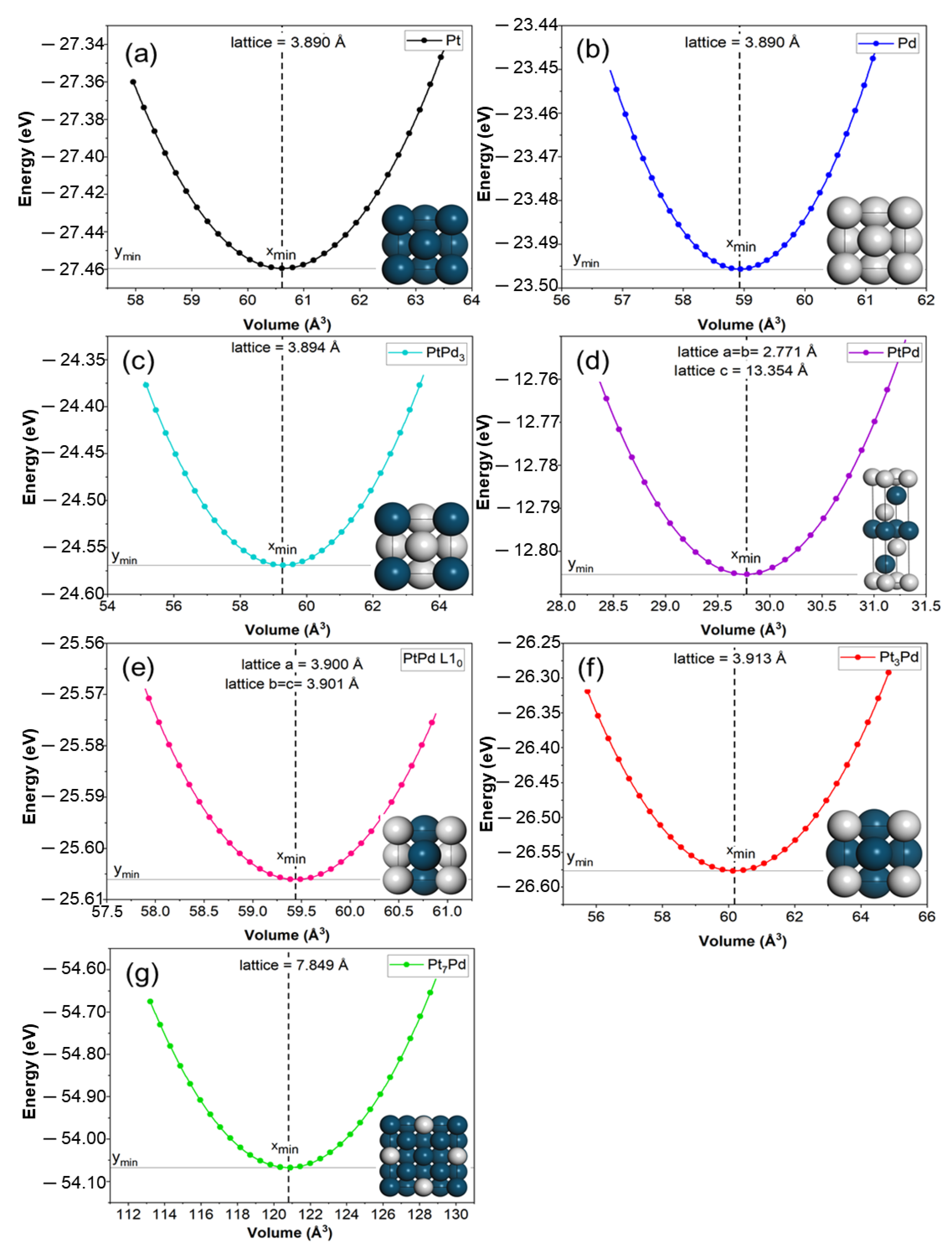
2.1. Structural Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit Cell Parameters | Unit Cell Angles | Equilibrium Volume | Space Group | Formation Energy | |||||
|---|---|---|---|---|---|---|---|---|---|
| (Å) | (°) | (Å3) | (no) | (eV/f.u) | |||||
| a | b | c | α | β | γ | ||||
| Pd | 3.890 | 3.89 | 3.890 | 90 | 90 | 90 | 58.86 | 225 | |
| Pd ref | 3.89 a,b | 3.890 | 3.89 | ||||||
| PtPd3 | 3.890 | 3.890 | 3.890 | 90 | 90 | 90 | 58.86 | 221 | −0.023 |
| PtPd3 ref | 3.88 c | 3.88 | 3.88 | 90 | 90 | 90 | |||
| PtPd | 2.771 | 2.771 | 13.354 | 90 | 90 | 120 | 88.80 | 166 | −0.050 |
| PtPd (L10) | 3.900 d | 3.901 d | 3.901 d | 90 d | 90 d | 90 d | 59.36 | 123 | −0.128 |
| Pt3Pd | 3.920 | 3.920 | 3.920 | 90 | 90 | 90 | 60.24 | 221 | −0.025 |
| Pt3Pd ref | 3.91c | 3.91 | 3.91 | 90 | 90 | 90 | |||
| Pt7Pd | 7.789 | 7.789 | 7.789 | 90 | 90 | 90 | 483.53 | 225 | −0.018 |
| Pt | 3.930 | 3.930 | 3.930 | 90 | 90 | 90 | 60.70 | 225 | |
| Pt ref | 3.92 b,c | 3.92 | 3.92 | ||||||
2.2. Elastic, Phonon, and Thermodynamic Properties
2.2.1. Elastic Properties
| Phase | C11 GPa | C12 GPa | C13 GPa | C14 GPa | C33 GPa | C44 GPa | B GPa | G GPa | E GPa | Ν GPa |
|---|---|---|---|---|---|---|---|---|---|---|
| Pt | 327.76 | 253.50 | 82.19 | 278 | 46–82 | 107–224 | 0.37–0.44 | |||
| Pt ref | 299 a, 238 b 250 c, 262 d | 46–54 e | 132–164 e | 0.48–0.51 e | ||||||
| Pd | 232.90 | 173.56 | 91.06 | 193 | 38–91 | 85–236 | 0.30–0.43 | |||
| Pd ref | 221 a 172 b 181 d | |||||||||
| PtPd3 | 256.55 | 183.97 | 86.67 | 216 | 47–90 | 107–237 | 0.32–0.42 | |||
| PtPd | 335.21 | 190.62 | 191.14 | 6.58 | 323.87 | 72.30 | 227–249 | 70–78 | 189–211 | 0.36–0.37 |
| 253 a | ||||||||||
| Pt3Pd | 327.20 | 215.01 | 91.72 | 217 | 45–72 | 108–193 | 0.35–0.42 | |||
| Pt7Pd | 372.38 | 210.00 | 73.76 | 264 | 74–81 | 202–221 | 0.36–0.37 |
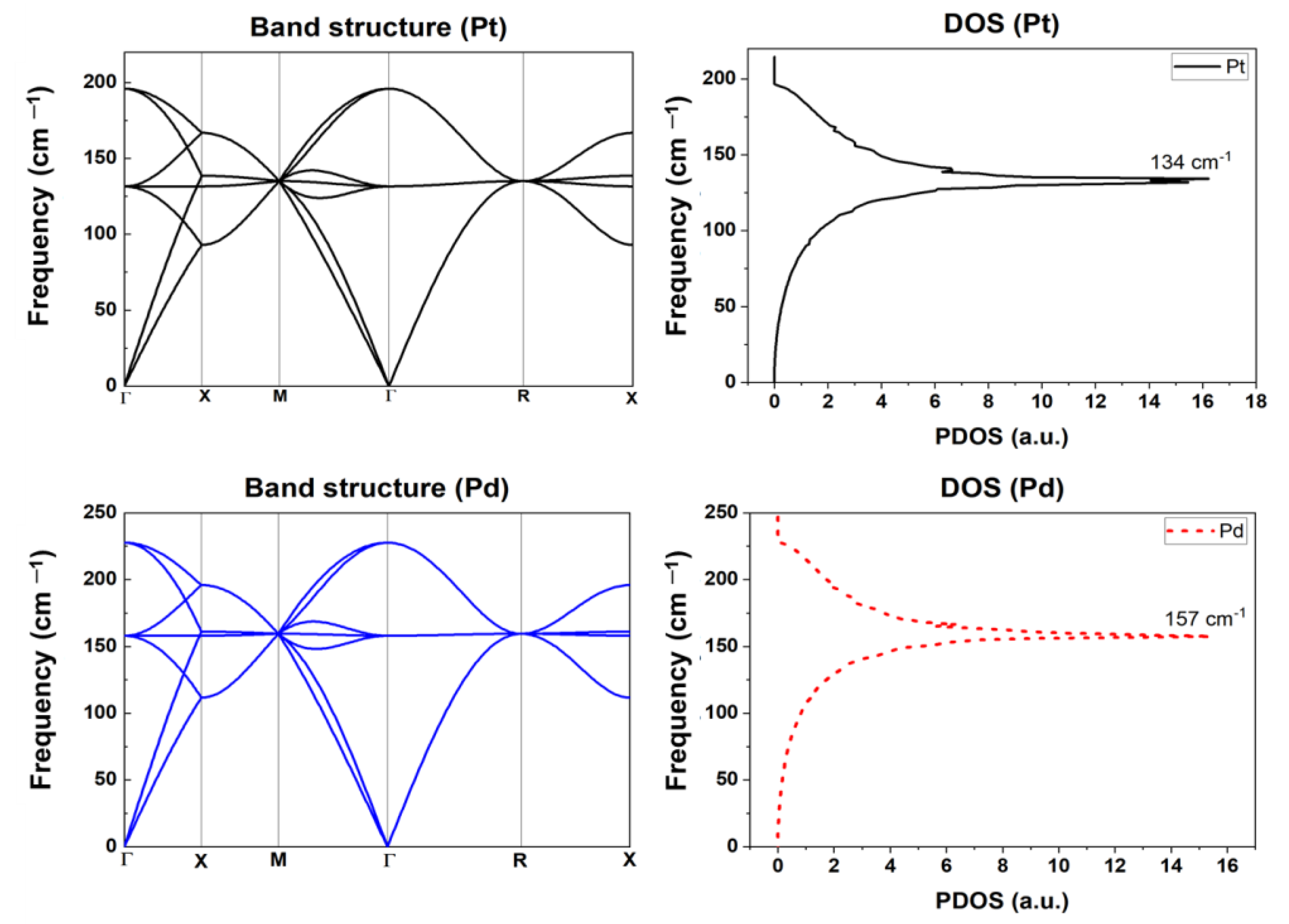
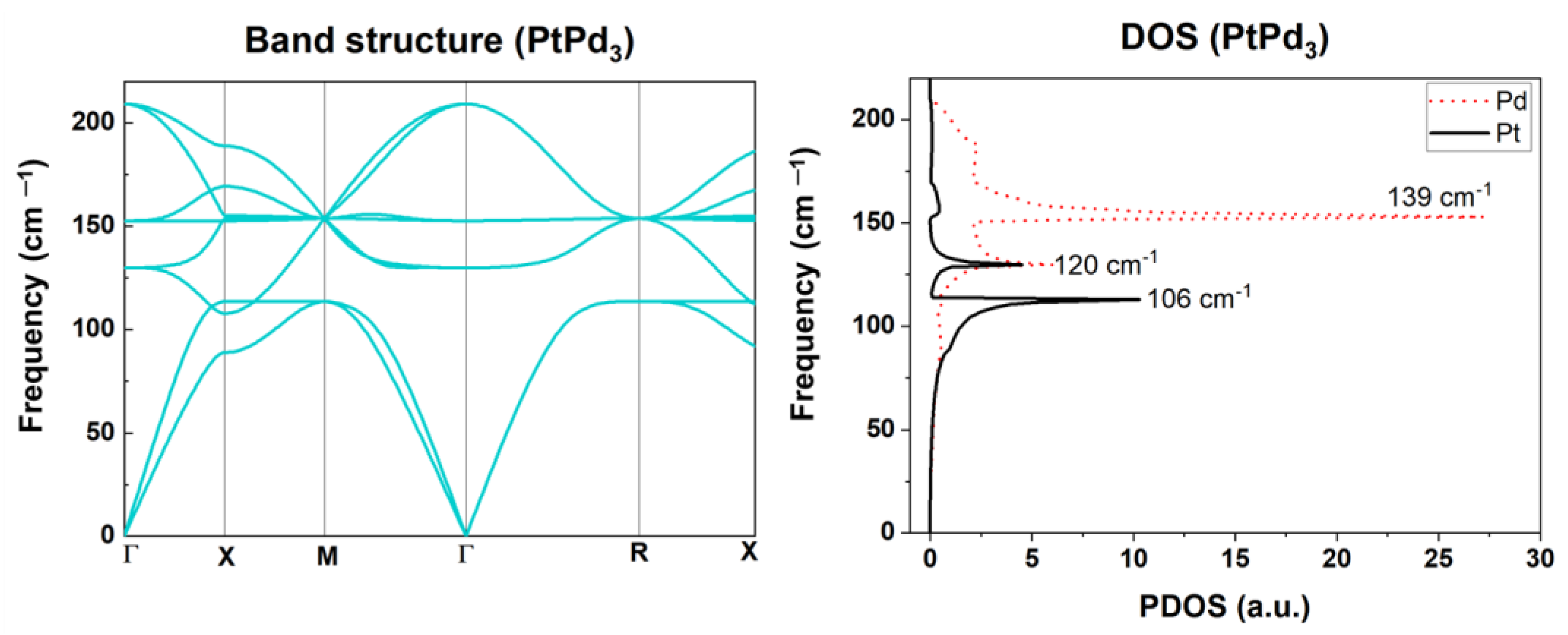
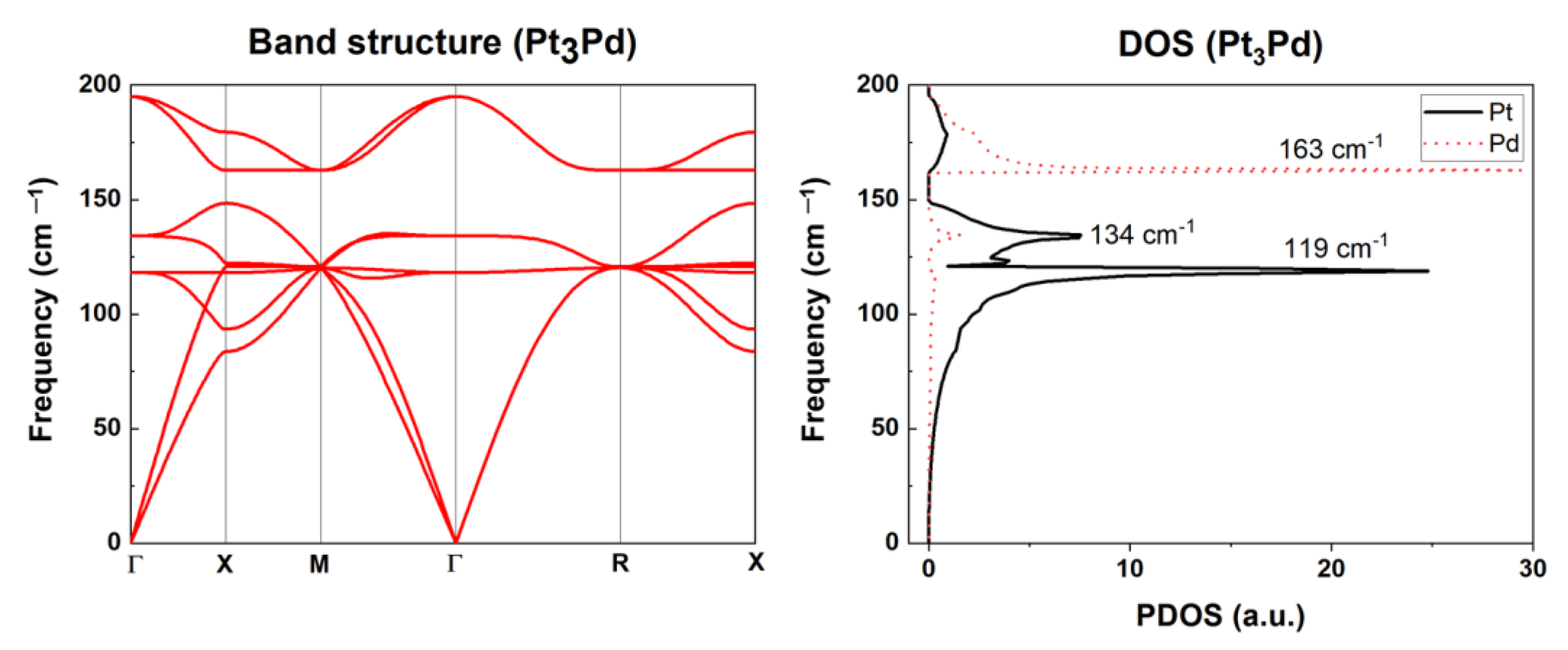
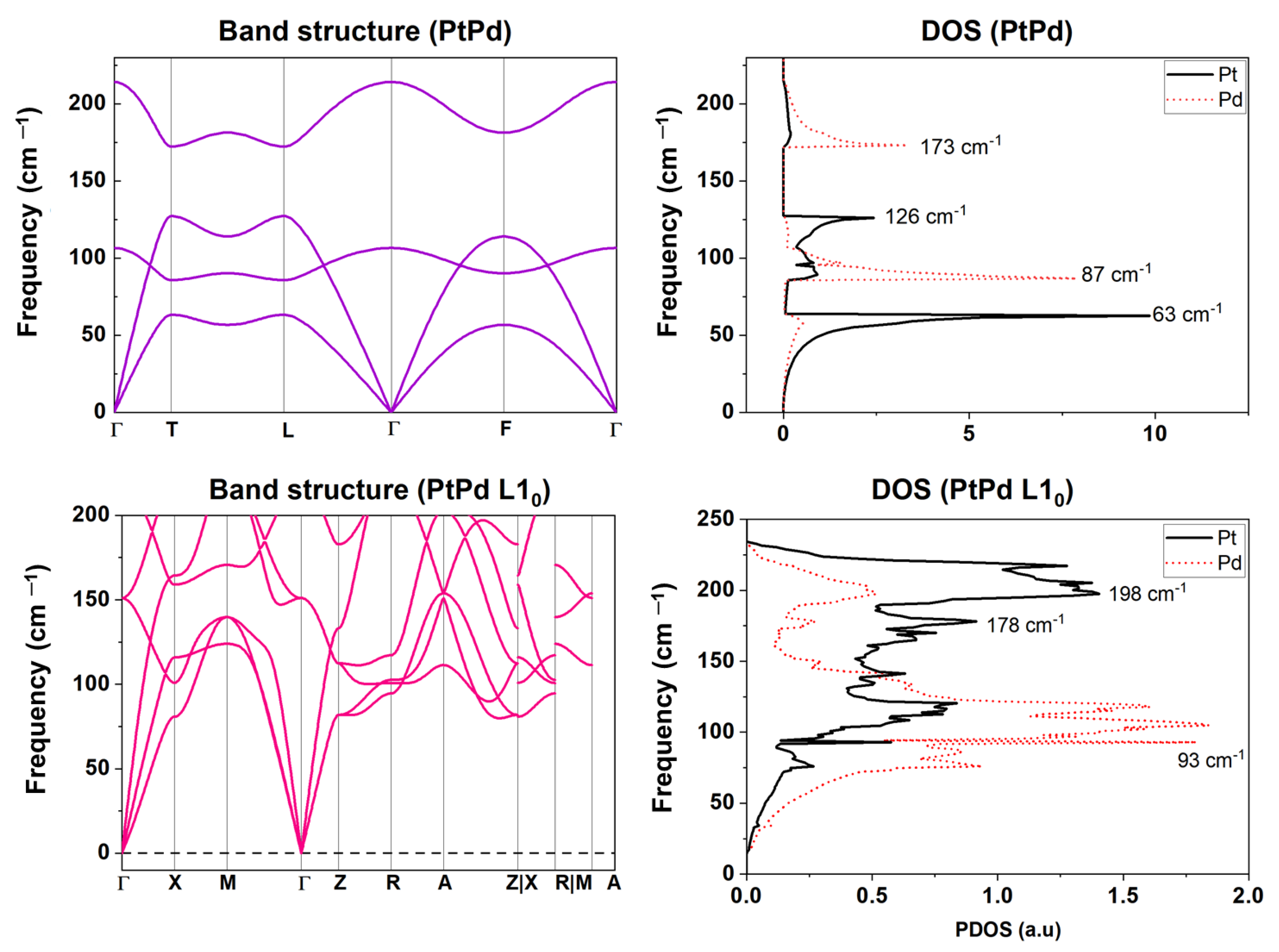
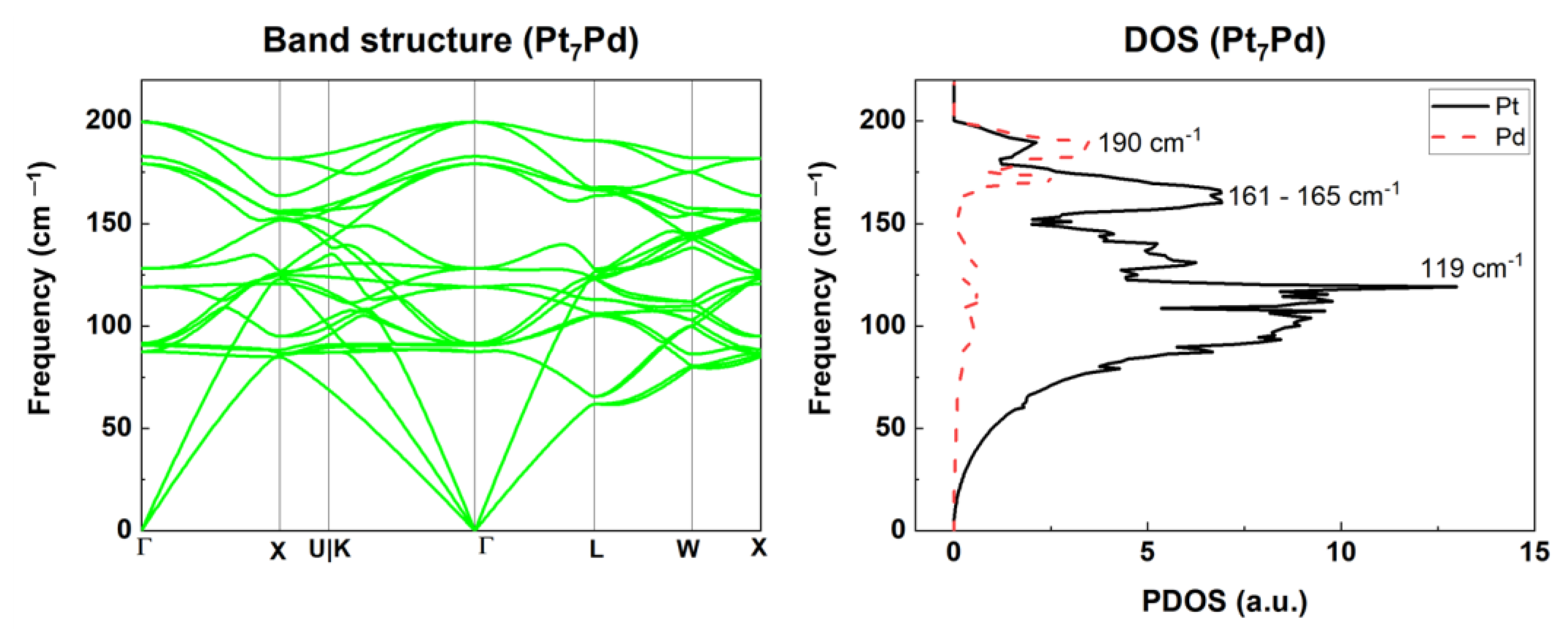
2.2.2. Phonon Properties
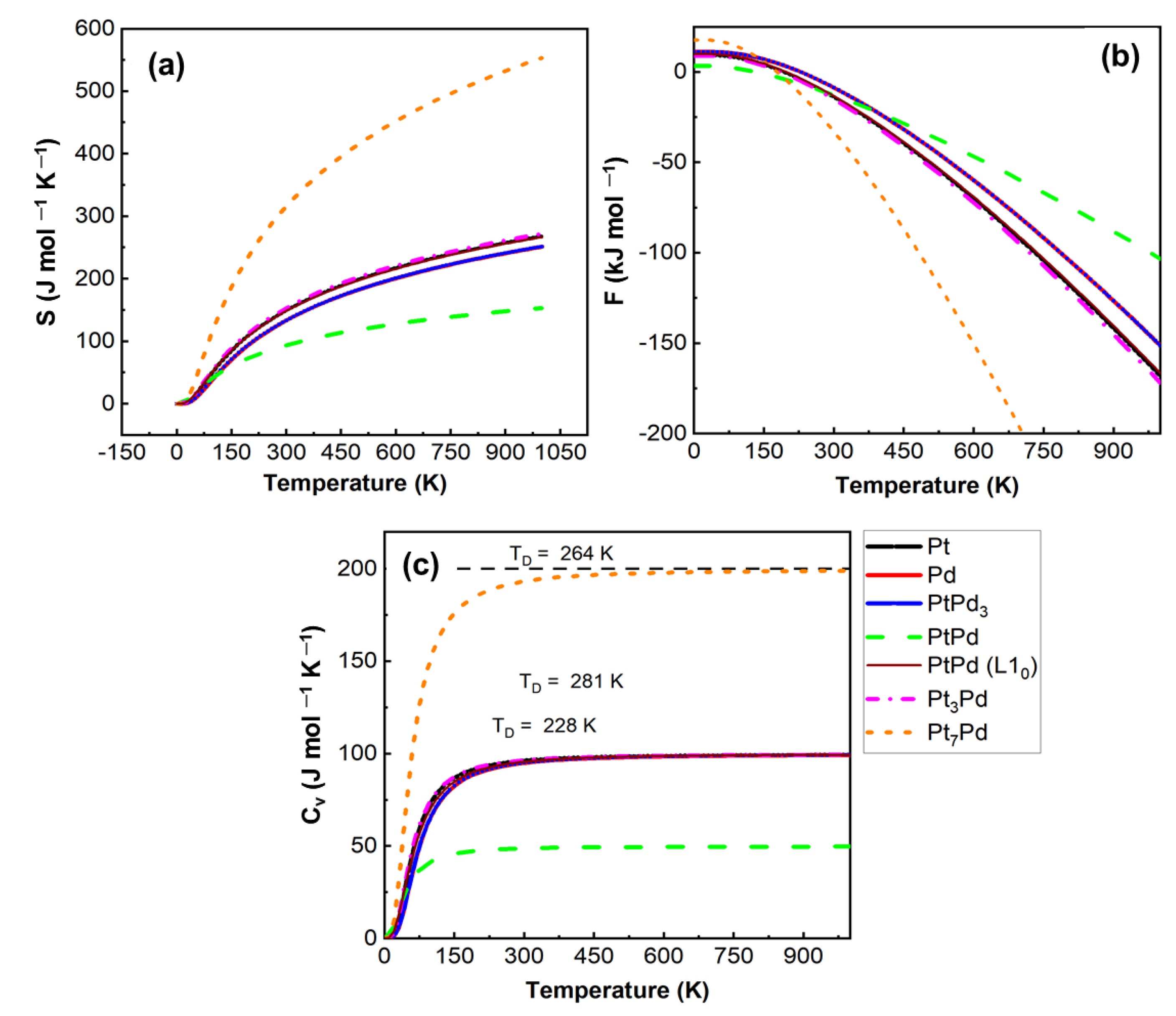
2.2.3. Thermodynamic Properties
2.3. Electronic Properties
3. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peake, S. Renewable Energy-Power for a Sustainable Future; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Elliott, D. Renewable energy and sustainable futures. Futures 2000, 32, 261–274. [Google Scholar] [CrossRef]
- Chi, J.; Yu, H. Water electrolysis based on renewable energy for hydrogen production. Chin. J. Catal. 2018, 39, 390–394. [Google Scholar] [CrossRef]
- Hirscher, M.; Yartys, V.A.; Baricco, M.; von Colbe, J.B.; Blanchard, D.; Bowman, R.C., Jr.; Broom, D.P.; Buckley, C.E.; Chang, F.; Chen, P.; et al. Materials for hydrogen-based energy storage–Past, recent progress and future outlook. J. Alloys Compd. 2020, 827, 153548. [Google Scholar] [CrossRef]
- Merker, J.; Lupton, D.; Topfer, M.; Knake, H. High temperature mechanical properties of the platinum group metals. Platin. Met. Rev. 2001, 45, 74–82. [Google Scholar]
- Abbasi, R.; Wu, L.; Wanke, S.; Hayes, R. Kinetics of methane combustion over Pt and Pt–Pd catalysts. Chem. Eng. Res. Des. 2012, 90, 1930–1942. [Google Scholar] [CrossRef]
- Pawelec, B.; Mariscal, R.; Navarro, R.; van Bokhorst, S.; Rojas, S.; Fierro, J. Hydrogenation of aromatics over supported Pt-Pd catalysts. Appl. Catal. A Gen. 2002, 225, 223–237. [Google Scholar] [CrossRef]
- Payot, F.; Reinecke, E.-A.; Morfin, F.; Sabroux, J.-C.; Meynet, N.; Bentaib, A.; March, P.; Zeyen, R. Understanding of the operation behaviour of a Passive Autocatalytic Recombiner (PAR) for hydrogen mitigation in realistic containment conditions during a severe Light Water nuclear Reactor (LWR) accident. Nucl. Eng. Des. 2012, 248, 178–196. [Google Scholar] [CrossRef]
- Screen, B.T. Platinum Group Metal Perovskite Catalysts. Platin. Met. Rev. 2007, 51, 87–92. [Google Scholar] [CrossRef]
- Cowley, A.; Woodward, B. A Healthy Future: Platinum in Medical Applications. Platin. Met. Rev. 2011, 55, 98–107. [Google Scholar] [CrossRef]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Obodo, K.O.; Ouma, C.N.M.; Bessarabov, D. Modified Pt (2 1 1) and (3 1 1) surfaces towards the dehydrogenation of methylcyclohexane to toluene: A density functional theory study. Appl. Surf. Sci. 2022, 584, 152590. [Google Scholar] [CrossRef]
- Duan, H.; Xu, C. Nanoporous PtPd Alloy Electrocatalysts with High Activity and Stability toward Oxygen Reduction Reaction. Electrochimica Acta 2015, 152, 417–424. [Google Scholar] [CrossRef]
- Briguglio, N.; Pantò, F.; Siracusano, S.; Aricò, A. Enhanced performance of a PtCo recombination catalyst for reducing the H2 concentration in the O2 stream of a PEM electrolysis cell in the presence of a thin membrane and a high differential pressure. Electrochimica Acta 2020, 344, 136153. [Google Scholar] [CrossRef]
- Du, L.; Prabhakaran, V.; Xie, X.; Park, S.; Wang, Y.; Shao, Y. Low-PGM and PGM-Free Catalysts for Proton Exchange Membrane Fuel Cells: Stability Challenges and Material Solutions. Adv. Mater. 2020, 33, 1908232. [Google Scholar] [CrossRef]
- Shao, Y.; Dodelet, J.; Wu, G.; Zelenay, P. PGM-Free Cathode Catalysts for PEM Fuel Cells: A Mini-Review on Stability Challenges. Adv. Mater. 2019, 31, e1807615. [Google Scholar] [CrossRef]
- Obodo, K.O.; Ouma, C.N.M.; Modisha, P.M.; Bessarabov, D. Density functional theory calculation of Ti3C2 MXene monolayer as catalytic support for platinum towards the dehydrogenation of methylcyclohexane. Appl. Surf. Sci. 2020, 529, 147186. [Google Scholar] [CrossRef]
- Lalik, E.; Kosydar, R.; Tokarz-Sobieraj, R.; Witko, M.; Szumełda, T.; Kołodziej, M.; Rojek, W.; Machej, T.; Bielańska, E.; Drelinkiewicz, A. Humidity induced deactivation of Al2O3 and SiO2 supported Pd, Pt, Pd-Pt catalysts in H2+O2 recombination reaction: The catalytic, microcalorimetric and DFT studies. Appl. Catal. A Gen. 2015, 501, 27–40. [Google Scholar] [CrossRef]
- Graham, G.W.; Jen, H.-W.; Ezekoye, O.; Kudla, R.J.; Chun, W.; Pan, X.Q.; McCabe, R.W. Effect of alloy composition on dispersion stability and catalytic activity for NO oxidation over alumina-supported Pt–Pd catalysts. Catal. Lett. 2007, 116, 1–8. [Google Scholar] [CrossRef]
- Morlang, A.; Neuhausen, U.; Klementiev, K.; Schütze, F.-W.; Miehe, G.; Fuess, H.; Lox, E. Bimetallic Pt/Pd diesel oxidation catalysts. Appl. Catal. B Environ. 2005, 60, 191–199. [Google Scholar] [CrossRef]
- Sanap, K.K.; Varma, S.; Waghmode, S.B.; Bharadwaj, S.R. Supported Pt nanoparticles for the hydrogen mitigation application. Int. J. Hydrogen Energy 2014, 39, 15142–15155. [Google Scholar] [CrossRef]
- Morfin, F.; Sabroux, J.-C.; Renouprez, A. Catalytic combustion of hydrogen for mitigating hydrogen risk in case of a severe accident in a nuclear power plant: Study of catalysts poisoning in a representative atmosphere. Appl. Catal. B Environ. 2004, 47, 47–58. [Google Scholar] [CrossRef]
- Li, Z.; Tian, B.; Zhen, W.; Wu, Y.; Lu, G. Inhibition of hydrogen and oxygen recombination using oxygen transfer reagent hemin chloride in Pt/TiO2 dispersion for photocatalytic hydrogen generation. Appl. Catal. B Environ. 2017, 203, 408–415. [Google Scholar] [CrossRef]
- Kalpakjian, S.; Schmid, S. Manufacturing, Engineering and Technology Si 6th Edition-Serope Kalpakjian and Stephen Schmid: Manufacturing, Engineering and Technology; Digital Designs: Oklahoma City, OK, USA, 2006. [Google Scholar]
- Meyers, M.A.; Chawla, K.K. Mechanical Behavior of Materials; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Mellor, B.G. Surface Coatings for Protection against Wear; Woodhead Publishing: Sawston, UK, 2006. [Google Scholar] [CrossRef]
- Hoornaert, T.; Hua, Z.K.; Zhang, J.H. Hard Wear-Resistant Coatings: A Review. In Advanced Tribology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 774–779. [Google Scholar] [CrossRef]
- Boyer, R.R.; Briggs, R.D. The Use of β Titanium Alloys in the Aerospace Industry. J. Mater. Eng. Perform. 2005, 14, 681–685. [Google Scholar] [CrossRef]
- Ahmed, S.; Zafar, M.; Shakil, M.; A Choudhary, M. Density functional theory study of structural, electronic, and thermal properties of Pt, Pd, Rh, Ir, Os and PtPd X (X = Ir, Os, and Rh) alloys. Chin. Phys. B 2016, 25, 036501. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef]
- Ou, L.; Chen, S. Comparative Study of Oxygen Reduction Reaction Mechanisms on the Pd(111) and Pt(111) Surfaces in Acid Medium by DFT. J. Phys. Chem. C 2013, 117, 1342–1349. [Google Scholar] [CrossRef]
- Darby, J.B.; Myles, K.M. A thermodynamic study of solid Pd-Pt alloys. Met. Trans. 1972, 3, 653–657. [Google Scholar] [CrossRef]
- Raub, E. Metals and alloys of the platinum group. J. Less Common. Met. 1959, 1, 3–18. [Google Scholar] [CrossRef]
- Kidron, A. An X-ray diffuse scattering measurement of local order in a Pd Pt alloys. Phys. Lett. A 1967, 25, 112–113. [Google Scholar] [CrossRef]
- Duan, Z.; Wang, G. Monte Carlo simulation of surface segregation phenomena in extended and nanoparticle surfaces of Pt–Pd alloys. J. Physics Condens. Matter 2011, 23, 475301. [Google Scholar] [CrossRef]
- Jacob, K.T.; Raj, S.; Rannesh, L. Vegard’s law: A fundamental relation or an approximation? Int. J. Mater. Res. 2007, 98, 776–779. [Google Scholar] [CrossRef]
- Woolley, J.C.; Berolo, O. Phase studies of the Pb1−xSnxSe alloys. Mater. Res. Bull. 1968, 3, 445–450. [Google Scholar] [CrossRef]
- Bis, R.F.; Dixon, J.R. Applicability of Vegard’s Law to the PbxSn1−xTe Alloy System. J. Appl. Phys. 1969, 40, 1918–1921. [Google Scholar] [CrossRef]
- Friedel, J. LX. Deviations from Vegard’s law, London, Edinburgh, Dublin Philos. Mag. J. Sci. 1955, 46, 514–516. [Google Scholar] [CrossRef]
- Murphy, S.T.; Chroneos, A.; Jiang, C.; Schwingenschlögl, U.; Grimes, R.W. Deviations from Vegard’s law in ternary III-V alloys. Phys. Rev. B 2010, 82, 073201. [Google Scholar] [CrossRef]
- Magomedov, M. On the deviation from the Vegard’s law for the solid solutions. Solid State Commun. 2020, 322, 114060. [Google Scholar] [CrossRef]
- Beldi, L.; Bendaoud, H.; Obodo, K.; Abbar, B.; Bouhafs, B. Density functional theory studies of the SrC and SrN compounds. Mater. Chem. Phys. 2019, 237, 121875. [Google Scholar] [CrossRef]
- Tang, K.; Wang, T.; Qi, W.; Li, Y. Debye temperature for binary alloys and its relationship with cohesive energy. Phys. B Condens. Matter 2018, 531, 95–101. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1954. [Google Scholar]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Budinski, K.G.; Budinski, M.K. Engineering Materials; Pearson Education India: Noida, India, 1999. [Google Scholar]
- Bozzolo, G.; Mosca, H.O.; del Grosso, M.F. Energy of formation, lattice parameter and bulk modulus of (Ni,X)Ti alloys with X = Fe, Pd, Pt, Au, Al, Cu, Zr, Hf. Intermetallics 2008, 16, 668–675. [Google Scholar] [CrossRef]
- Bercegeay, C.; Bernard, S. First-principles equations of state and elastic properties of seven metals. Phys. Rev. B 2005, 72, 214101. [Google Scholar] [CrossRef]
- Dieter, G.E.; Bacon, D. Mechanical Metallurgy; McGraw-Hill: New York, NY, USA, 1976. [Google Scholar]
- Köster, W.; Franz, H. Poisson’s ratio for metals and alloys. Metall. Rev. 1961, 6, 1–56. [Google Scholar] [CrossRef]
- Haines, J.; Léger, J.; Bocquillon, G. Synthesis and Design of Superhard Materials. Annu. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]
- Cao, D. Pair Distribution Functions. In Molecular Dynamics Simulations of Interfaces; West Virginia University: Morgantown, WV, USA, 2006. [Google Scholar]
- Toby, B.H.; Egami, T. Accuracy of pair distribution function analysis applied to crystalline and non-crystalline materials. Acta Crystallogr. Sect. A Found. Crystallogr. 1992, 48, 336–346. [Google Scholar] [CrossRef]
- Benaissa, H.; Benatmane, S.; Amari, S.; Obodo, K.O.; Beldi, L.; Bendaoud, H.; Bouhafs, B. Ferromagnetism in RaBi with Zinc-Blende and Wurtzite Structures: Ab-initio Prediction. SPIN 2018, 08, 1850008. [Google Scholar] [CrossRef]
- Beldi, L.; Bendaoud, H.; Obodo, K.O.; Abbar, B.; Bouhafs, B. Prediction of a Dynamically Stable New Half-Metallic Phase for the BaN and BaC Compounds. J. Supercond. Nov. Magn. 2018, 32, 2031–2044. [Google Scholar] [CrossRef]
- Cahn, R.W. Binary Alloy Phase Diagrams, 2nd ed.; ASM International: Materials Park, OH, USA, 1991. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Kerkar, A.; Tripathi, S.; Dharwadkar, S. The palladium-platinum phase diagram. J. Less Common Met. 1991, 169, 167–172. [Google Scholar] [CrossRef]
- Guisbiers, G.; Abudukelimu, G.; Hourlier, D. Size-dependent catalytic and melting properties of platinum-palladium nanoparticles. Nanoscale Res. Lett. 2011, 6, 396. [Google Scholar] [CrossRef]
- Atkins, P.; De Paula, J.; Keeler, J. Atkins’ Physical Chemistry International Edition, 11th ed.; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Botha, L.M.; Santos-Carballal, D.; Terranova, U.; Quesne, M.G.; Ungerer, M.J.; van Sittert, C.G.C.E.; de Leeuw, N.H. Mixing thermodynamics and electronic structure of the Pt1−xNix (0 ≤ x ≤ 1) bimetallic alloy. RSC Adv. 2019, 9, 16948–16954. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C.M. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. npj Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. Spglib: A software library for crystal symmetry search. arXiv 2018, arXiv:1808.01590. [Google Scholar]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-General Least-Squares Extraction of Elastic Coefficients from Ab Initio Total Energy Calculations. Phys. Rev. B 2001, 63, 174103. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-General Least-Squares Extraction of Elastic Data for Strained Materials from Ab Initio Calculations, of Stress. Phys. Rev. B 2002, 65, 104104. [Google Scholar] [CrossRef]
- Anderson, O.L. A Simplified Method for Calculating the Debye Temperature from Elastic Constants. J. Phys. Chem. Solids 1963, 24, 909. [Google Scholar] [CrossRef]
- Grüneisen’s, E. Zustand des festen Körpers. In Thermische Eigenschaften der Stoffe; Handbuch Der Physik; Springer: Berlin/Heidelberg, Germany, 1928. [Google Scholar]
- Mayer, B.; Anton, H.; Bott, E.; Methfessel, M.; Sticht, J.; Harris, J.; Schmidt, P.C. Ab-Initio Calculation of the Elastic Constants and Thermal Expansion Coefficients of Laves Phases. Intermetallics 2003, 11, 23–32. [Google Scholar] [CrossRef]

| Structure | Atom | q (e/atom) |
|---|---|---|
| Pt | Pt | 0.00 |
| Pd | Pd | 0.00 |
| PtPd3 | Pt | −0.10 |
| Pd | 0.03 | |
| PtPd | Pt | −0.11 |
| Pd | 0.11 | |
| Pt3Pd | Pt | −0.03 |
| Pd | 0.09 | |
| Pt7Pd | Pt | −0.03 |
| Pd | 0.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botha, L.M.; Ouma, C.N.M.; Obodo, K.O.; Bessarabov, D.G.; Sharypin, D.L.; Varyushin, P.S.; Plastinina, E.I. Ab Initio Study of Structural, Electronic, and Thermal Properties of Pt/Pd-Based Alloys. Condens. Matter 2023, 8, 76. https://doi.org/10.3390/condmat8030076
Botha LM, Ouma CNM, Obodo KO, Bessarabov DG, Sharypin DL, Varyushin PS, Plastinina EI. Ab Initio Study of Structural, Electronic, and Thermal Properties of Pt/Pd-Based Alloys. Condensed Matter. 2023; 8(3):76. https://doi.org/10.3390/condmat8030076
Chicago/Turabian StyleBotha, Louise Magdalene, Cecil Naphtaly Moro Ouma, Kingsley Onyebuchi Obodo, Dmitri Georgievich Bessarabov, Denis Lvovich Sharypin, Pyotr Sergeevich Varyushin, and Elizaveta Ivanovna Plastinina. 2023. "Ab Initio Study of Structural, Electronic, and Thermal Properties of Pt/Pd-Based Alloys" Condensed Matter 8, no. 3: 76. https://doi.org/10.3390/condmat8030076
APA StyleBotha, L. M., Ouma, C. N. M., Obodo, K. O., Bessarabov, D. G., Sharypin, D. L., Varyushin, P. S., & Plastinina, E. I. (2023). Ab Initio Study of Structural, Electronic, and Thermal Properties of Pt/Pd-Based Alloys. Condensed Matter, 8(3), 76. https://doi.org/10.3390/condmat8030076