The reference part is purely repulsive and it determines the structure of dense liquids, while the attractive perturbing component is responsible for cohesion and phase changes [
10]. A coupling-parameter
is introduced into the potential and it is expressed as
. Its magnitude
determines the strength of the perturbation; with
being the reference part and
the full potential. All of the correlation functions, which determine the structure of the fluid, like the pair distribution function
, are now to be treated as functions of
. In the thermodynamic perturbation theory (TPT), these functions are expanded as Taylor’s series in
regarding their reference part as
Here,
is the reference system’s pair distribution function and
denotes its
derivatives at
. Similar Taylor’s series representations hold for other correlation functions as well: the direct correlation function
, the total correlation function
, and the indirect correlation function
. For all of them, the subscript 0 will denote the function that corresponds to the reference system. The effect of the perturbation is accurately determined if the derivatives, like
, are obtained up to sufficient orders. Traditional perturbation theory is mainly limited to the first two orders [
7]; however, CPE accounts for quite higher orders by taking recourse to the OZE, which defines the relation between the short ranged direct correlation function
and the long ranged total correlation function
. The latter is the appropriate function to be used in the OZE as it tends to zero for large
r, just like the pair-potential. For one-component systems with spherically symmetric potential, as in the present case, the OZE is written as:
where
is the number density of atoms. This relation has to be supplemented with a closure relation involving
,
and the pair-potential
. An exact closure is unfeasible; however, several approximate forms are available [
3]. All of the closures are generally expressed in terms of a ’bridge function’
in the form:
where
,
Boltzmann’s constant and
T absolute temperature. Note that
is also scaled with energy unit
. Approximate forms of
generate different closures; for instance, the one called HNC that was mentioned earlier corresponds to the choice
.
A ’bar’ is used throughout to denote Fourier transforms. It is useful to rewrite these equations in terms of the functions and and the symmetric kernel , as discussed below.
The two algebraic equations Equations (
17) and (
18), although defined in two different spaces, define the non-linear systems that determine the correlation functions and entire thermodynamic properties. When the inter-particle potential contains an attractive component, solutions to the OZE with most of the closures develop singularities in the liquid-vapor transition region [
14]. Hence, the determination of the critical points and phase diagrams is a difficult task, and very careful strategies need to be implemented [
27]. The CPE technique completely avoids this problem of dealing with singularities, as all correlation functions are expanded in Taylor’s series. The issues that are related to convergence of the series and ’no-solution-region’ are discussed later. The Newton–Armijo non-linear solver considered next is applicable to general potentials; however, it is discussed below for the case of reference potential, as required in the CPE method.
5.1. Newton-Armijo Solver for Reference System
For repulsive inter-particle potentials, as in the case of reference system, an accurate form of bridge function [
28] is
, so the closure is expressed as
The solution to the OZE in terms of
is given by
These nonlinear equations are readily solved while using Newton’s method [
29]. While, in this reference, these are treated as two equations and two unknowns, it is possible to take Equation (
20) as providing
if
is given. Accordingly, only Equation (
21) is to be solved by Newtons’ method. Starting with initial guess solution
(and, hence,
,
and
), an improved solution
is obtained while using the first order correction terms:
where
is the derivative with respect to
. Taking Fourier transform of
and substituting in the second equation yields
The linear operator
defining this equation involves the following operations: (i) Fourier inverse taking
to
, (ii) multiplication by
, (iii) Fourier transform of the resulting product, and (iv) multiplication by
. This may be symbolically written as:
, where
denotes the Fourier transform. Thus, the action of
essentially involves two Fourier transform operations, which is re-defined with a symmetric kernel as:
where
and
. Subsequently, the operator
is re-defined as
, where
represents integration with the symmetric kernel
. The adjoint of
, as needed in Krylov space-based linear equation solvers discussed next, is then easily computed as
.
Numerical implementation of this algorithm uses a uniformly spaced discrete coordinates
with
where
is, typically, 10. This choice allows the use of FFT (Fast Fourier-transform) techniques. With a typical
, the discrete mesh sufficiently extends to account for the asymptotic variation of the correlation functions. In Fourier space,
is chosen, such that
, so that orthogonality of trigonometric functions is maintained in the discrete space. With these choices, the linear equation for
is reduced to a matrix equation of order
, which is most efficiently solved while using Krylov space-based methods [
12].
The process of improving the solution, called Newton’s iterations, is continued until the Euclidean norm is less than a prescribed value. If the norms satisfy the condition (subscripts ’1’ and ’0’ denote the current and previous iteration values, and is a small number), next Newton’s iteration follows. Otherwise, it is likely that iteration with full step would diverge. Accordingly, a smaller step size with is to be used. Note that the norms correspond to , while to . Armijo’s rule restricts the new step size to with a value of . For choosing that , a sub-iteration yielding with is performed. A quadratic fit of versus is then used to obtain , which corresponds to the minimum of in the interval . Another sub-iteration providing is performed and, if it satisfies the norm-reduction criterion, next Newton’s iteration follows. Otherwise, more sub-iterations are performed, each time with the replacements and , and, similarly, the respective norms. If the sub-iterations exceed a prescribed limit 10), it would be necessary to restart Newton’s iterations with a new guess solution.
The linear equations to be solved, at each Newton’s iteration, is of the standard form
. The Conjugate Gradient (CG) method is the simplest of all iterative methods attempting a solution in the Krylov space, although it is applicable to symmetric matrices [
30]. For non-symmetric matrices, as in the present case, the CG method is applicable to the normal equation
as
is symmetric. The algorithm starts with a guess solution
and its error vector
, and auxiliary vectors
. Afterwards, the iteration process, called CGNR (Conjugate Gradient to Normal equations to minimize Residual) (see
Appendix A), generates new solutions
, such that the residual error norm
is minimized at each iteration
n. The process converges to the exact solution in utmost
(dimension of the matrix) iterations if
is non-singular [
30]. In practice, iterations are terminated when the error norm
satisfies a prescribed criteria. There is little point in getting a high degree of convergence in the early stages of Newton’s iterations. In what are called inexact methods, the criteria used are
with a typical value
. A more common method, called GMRES (Generalized Minimum Residual), although it does not need
, requires much larger storage space [
30].
The correlation functions of the reference system are determined with the methods discussed (also see
Appendix A) in the entire fluid-phase-plane, except at very low temperatures, where perturbation theories are inapplicable.
5.2. Derivatives of Correlation Functions
The next step in CPE is to compute the derivatives, like
in Equation (
15). Writing the general closure relation in Equation (
17) in the form
, where
, repeated differentiation with respect to
yields
where
denote the Binomial coefficients and
is the Kroenecker delta. This is valid for arbitrary
and, in particular, for
. The arguments
r and
of all functions are omitted for simplifying the equations. Note that
is the attractive component in the potential according to the WCA prescription. The summation in Equation (
25) only contains derivatives lower than
n as the term containing
is separated out. Substituting for
and using the relation
for
yields the recursion relation:
The derivative
is written as
, so that
only contains derivatives of
y of an order lower than
n. This is needed, as
is yet to be determined with another equation. The definition of structure factor:
shows that
for
. From now on, the arguments
k and
of all Fourier functions are also omitted. Repeated differentiation of the relation
yields
where the first and last terms are separately written from the summation. Simplifying the expression and substituting in
gives the recursion
Here, the definition of
is used and
is substituted as
in the summation. Note that
only contains lower order derivatives
and
for
. Taking Fourier transform of Equation (
26) and substituting in Equation (
29) yields a linear equation for
, which is expressed as
This expression is valid for arbitrary value of
. The linear operator in this equation (for all
n) is identical to that shown in Equation (
23) if the derivatives are evaluated at
, which is, for the reference system. Thus, the Krylov space-based methods mentioned above are also applicable here; with the only change being the source term
.
5.4. Thermodynamic Functions
After computing the Taylor’s series for all functions to an arbitrary high order, setting
provides correlation functions that correspond to the full potential. The results that are discussed below are obtained with 7
th order approximation, as a further increase in order does not have any significant effect. Thermodynamic properties, like pressure
and inverse compressibility
, are expressed as
Similar expressions hold for internal energy and chemical potential [
27]. The argument
is omitted from these expressions. For computational purposes, Equation (
33) is re-expressed in terms of the ’cavity function’
, as:
Here,
is the second virial coefficient for
U, and it is separated out explicitly. An excellent first order perturbation theory [
31] for the equation of state follows from this equation with the approximations:
and
, thereby terminating the integral at
. The second approximation is the basis of first order theory. The first approximation is motivated from the observation that derivative
is only predominant near
, where
is small.
The OZE does not provide exact thermodynamic consistency [
3] in the sense that pressure that is computed from different routes does not agree with each other. The inverse compressibility
, being expressed in terms of the short ranged correlation function
, is the more appropriate response function to compute pressure via integration with respect to
. The method followed here employs a fine mesh of
T and
over the phase plane where
is computed. Subsequently, two-dimensional (2-D) interpolation is used for intermediate values, and
P is obtained via integration over
from zero. Solutions of the critical conditions
provide the critical point parameters and Maxwell’s construction the phase diagram. Using thermodynamic relations to obtain other quantities, like energy and entropy, equation of state tables [
32] can be generated in the expanded volume regions.
There are some important issues underlying the above procedure. It is known that there are multiple solutions to OZE and closure relations in the two-phase region when attractive interactions are present. This is illustrated in Baxter’s analytical solution [
33] of the sticky hard sphere model. These include isotherms with negative compressibility, complex, and even diverging pair distribution function. Numerical methods also show similar features for general potentials and closure relations (see following section). Solutions that are given by van der Waals equation, first order perturbation theory, and the CPE show isotherms with negative compressibility in the two-phase region. The nonphysical pressure
that is generated by these approaches is replaced with a flat isotherm while using thermodynamic conditions of equal pressure and chemical potential, or the equivalent Maxwell’s construction. The use of virial pressure (Equation (
33)) or inverse compressibility (Equation (
34)) produces similar results via Maxwell’s construction, although some differences arise due to the inherent thermodynamic inconsistency mentioned earlier. Another approach is to compute virial pressure and chemical potential, only in regions where compressibility is positive, and then derive the phase diagram while using thermodynamic conditions [
27]. The structure factor
within the two-phase region is to be obtained as a weighted average of contributions of the gas and liquid phases that correspond to any point on the isotherm. Only renormalization group approaches, such as in the the hierarchical reference theory [
34], generate flat isotherms in the two-phase region without using Maxwell’s construction.
Yet another relevant issue regarding the utility of Taylor’s series expansion of (and other correlation functions) needs clarification. Negative compressibility, at any phase point, implies that is negative and, by continuity requirements, would vanish at some , as it should approach unity for large k. Therefore, and, consequently, would diverge at . Thus, negative compressibility at any phase point implies a singularity in the Fourier transform (The author thanks one of the anonymous reviewers of Condensed Matter (MDPI) journal for this argument). Taylor’s series expansions that are assumed in CPE do not converge to this singular solution, in fact, numerical results indicate that they converge to a different solution (see following section). This is plausible in a scenario, wherein multiple solutions exist in the two-phase region. Their utility in computing phase diagram (via thermodynamic or Maxwell’s construction) needs to be evaluated by comparing the results with those from independent methods.
5.5. ’No-Solution-Region ’
Multiple solutions and singularities of the OZE in the liquid-vapor transition region have been investigated in detail for the Lennard-Jones (LJ) potential:
, with the HNC closure [
13] and other closures [
14]. The pseudo-arc-length continuation algorithm is needed for the direct application of Newton’s method to the full potential for tracking these singularities [
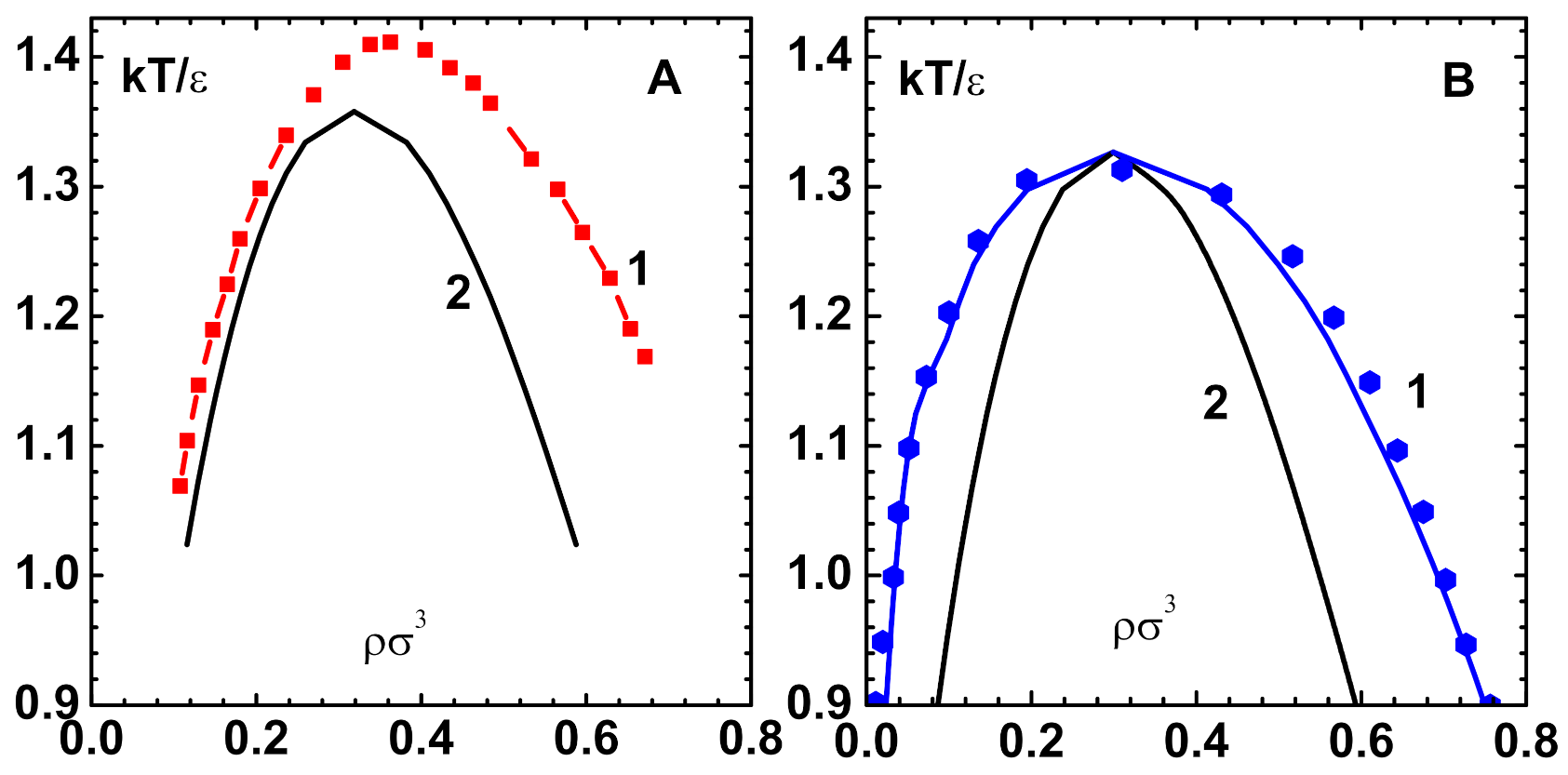
13]. The no-solution-line is defined as the curve in the phase plane, along which the Jacobian is singular and so Newton’s method is inapplicable.
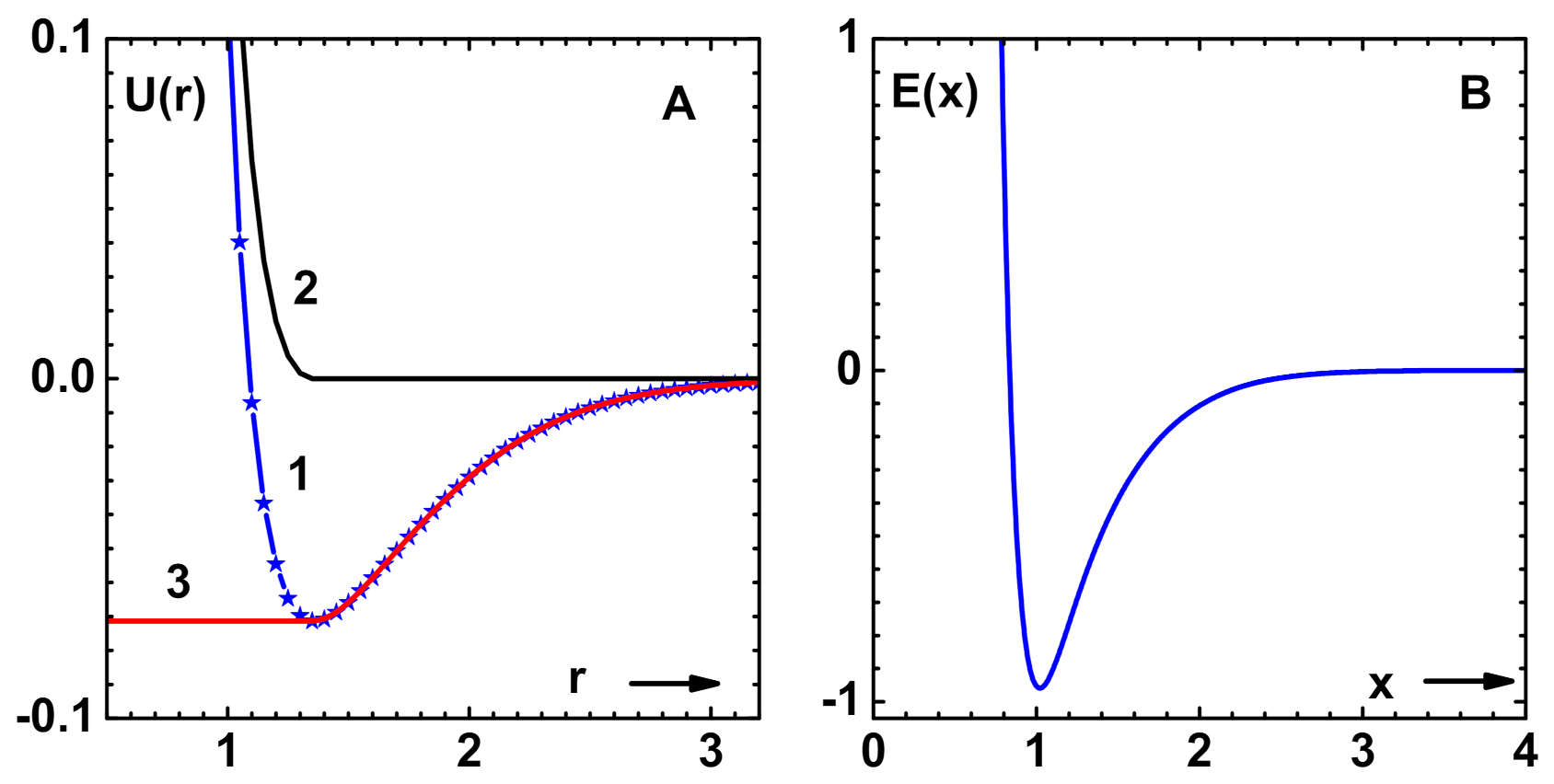
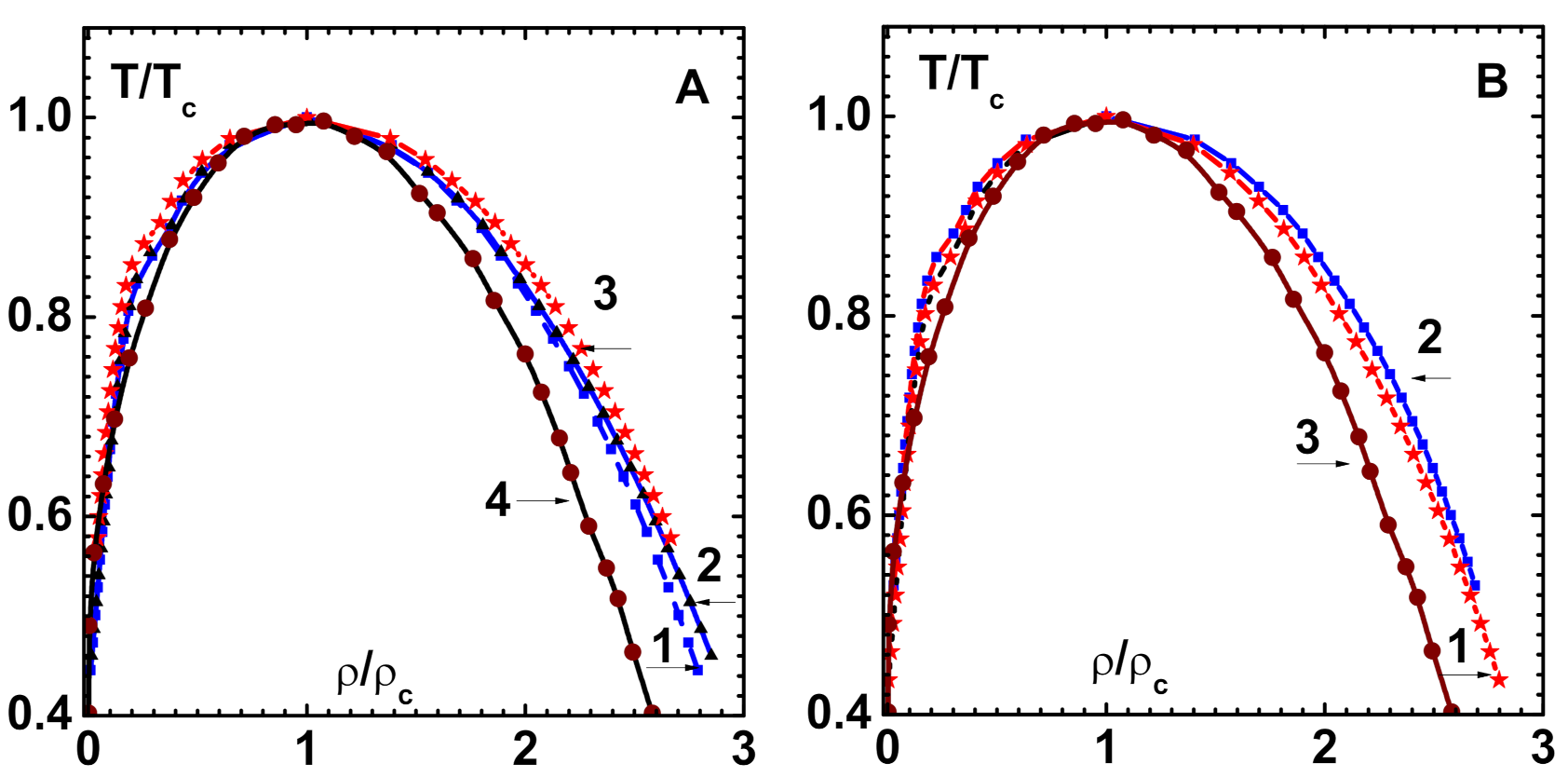
Figure 3A displays (curve-1) this curve for the LJ-HNC model [
13].
In fact, there are more characteristic features, like the ’fold-bifurcation’ of solutions in the liquid-vapor region of the nonlinear OZE. However, the CPE method does not pick up these singularities, as it is based on series expansion about the repulsive component of the potential. The situation is similar that of van der Waals equation, although free of singularities, which predicts a region of negative compressibility.
Figure 3A also shows the spinodal line ( defined by
) for the LJ-HNC model, which is obtained using 7th order CPE of this paper (curve-2).
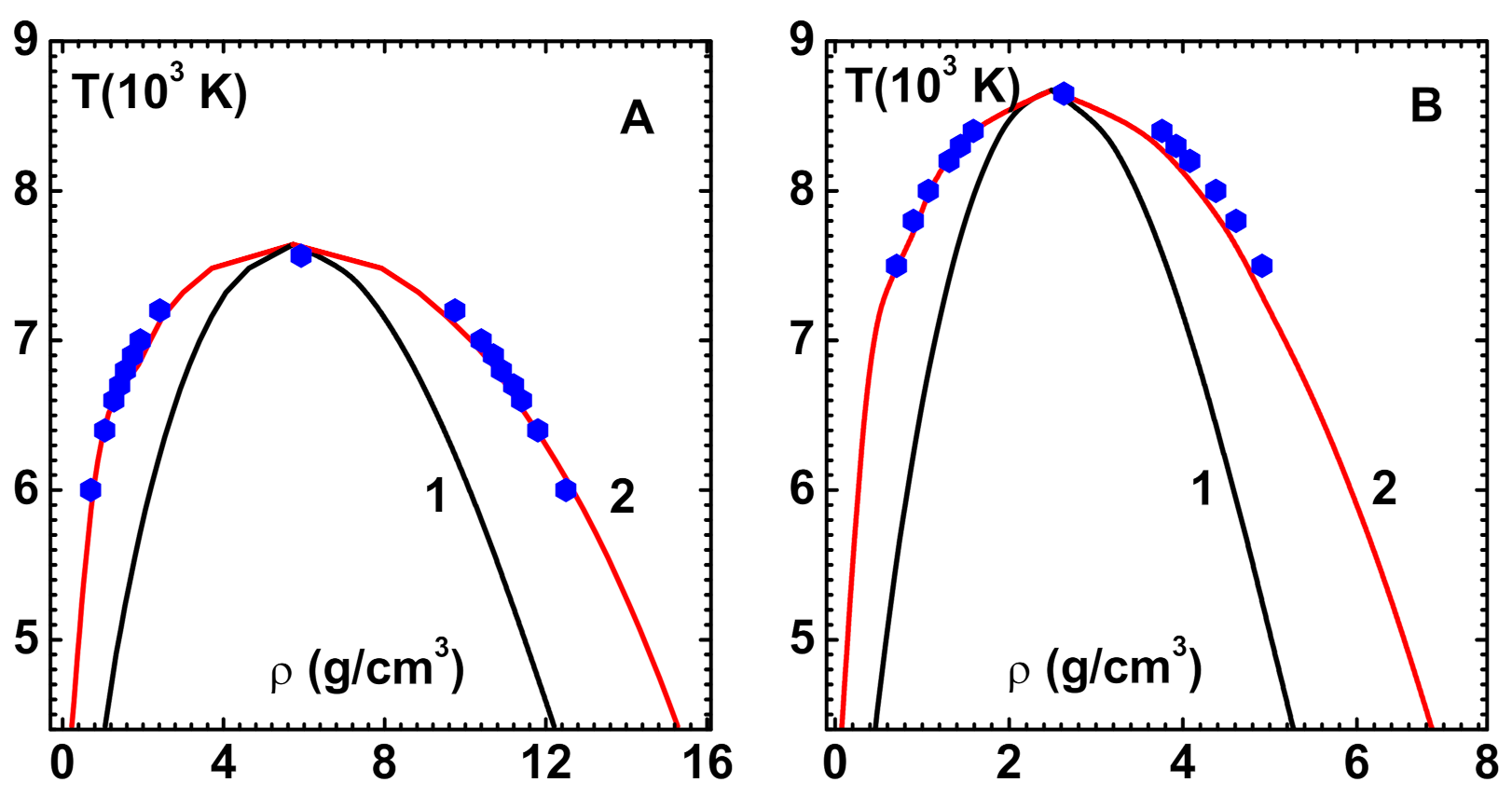
The LJ system is also investigated while using the density-dependent bridge function [
28] for comparison with simulation data [
35]. This is important because the bridge function is the sole ingredient that determines the predictive power of OZE. The results for phase diagram (co-existence curve) that were obtained with CPE shown (curve-1) in
Figure 3B compare well with simulation data (symbols). There is only slight disagreement along the liquid branch near the transition point. The critical point parameters obtained:
and
also compare well with simulation data given in square brackets. The spinodal line (curve-2), which is different from that obtained with HNC, is also shown.
As discussed in the previous section, it is necessary to check wheter the CPE generates any meaningful correlation functions, particularly in the two-phase region.
Figure 4A shows the direct correlation function
(curve-c), the reference function
(curve-0), and six successive derivative terms
(curve-1 to curve-6) for the LJ system using the density-dependent bridge function [
28]. These correspond to the phase point
and
, which is in the spinodal region. It is evident that the expansion approaches a well defined function
. The reference function
is mostly negative, but the positive part in
for
, arising out of attractive interactions, gives rise to negative compressibility. Similarly,
Figure 4B shows the pair distribution function
(curve-g), the reference function
(curve-0), and six derivative terms
(curve-1 to curve-6), also corresponding to the same phase point. The reference function
with a small peak develops to
with two well defined peaks due to attractive interactions. It will be more accurate to use the series expansions in Equation (
17) (closure relation) for computing
. These ’correlation functions’ are nonphysical, as they generate negative compressibility and van der Waals’s loop in pressure, but they may be used in order to compute phase diagram via Maxwell’s construction. Afterwards, it will be necessary to take the weighted averages (as explained earlier for structure factor) of contributions of gas and liquid phases to obtain physically acceptable correlation functions.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}