

Hidden Compositional Heterogeneity of Fish Chromosomes in the Era of Polished Genome Assemblies



Abstract
1. Introduction
2. Materials and Methods
2.1. Code and Availability
2.2. Genome Assemblies Used, Their History, and the Underlying Sequencing
2.3. Soft-Masking of Genome Assemblies
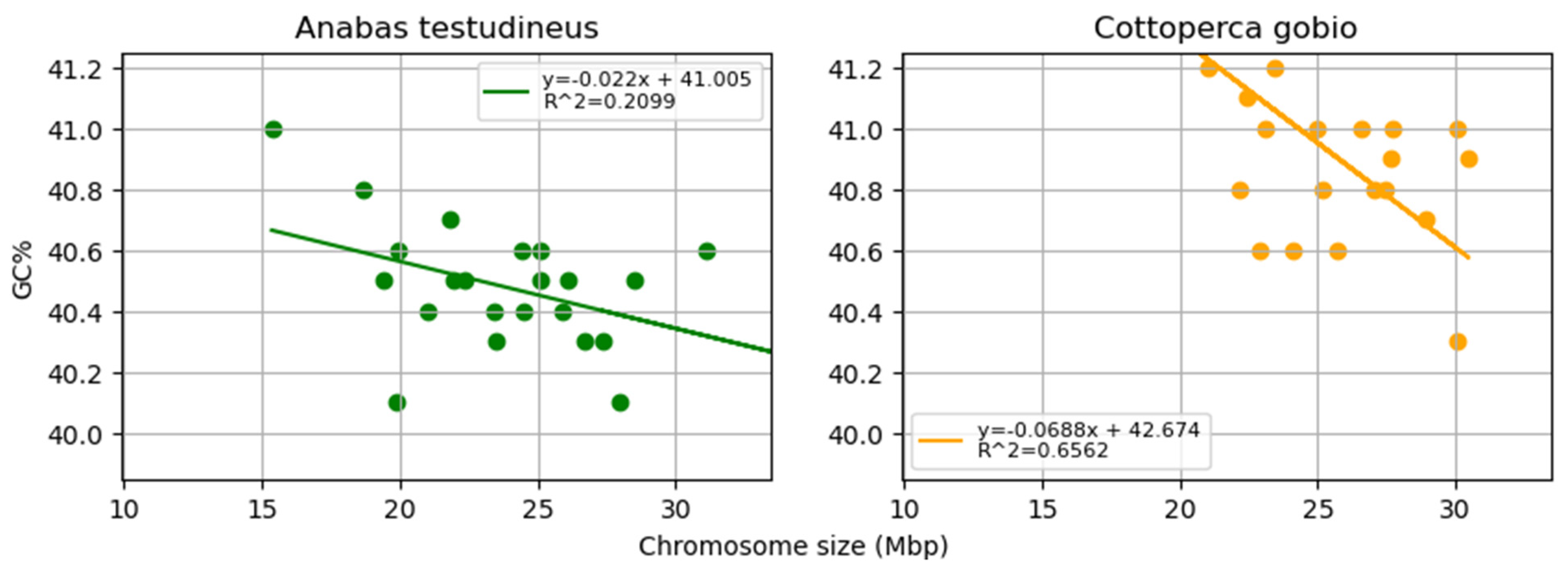
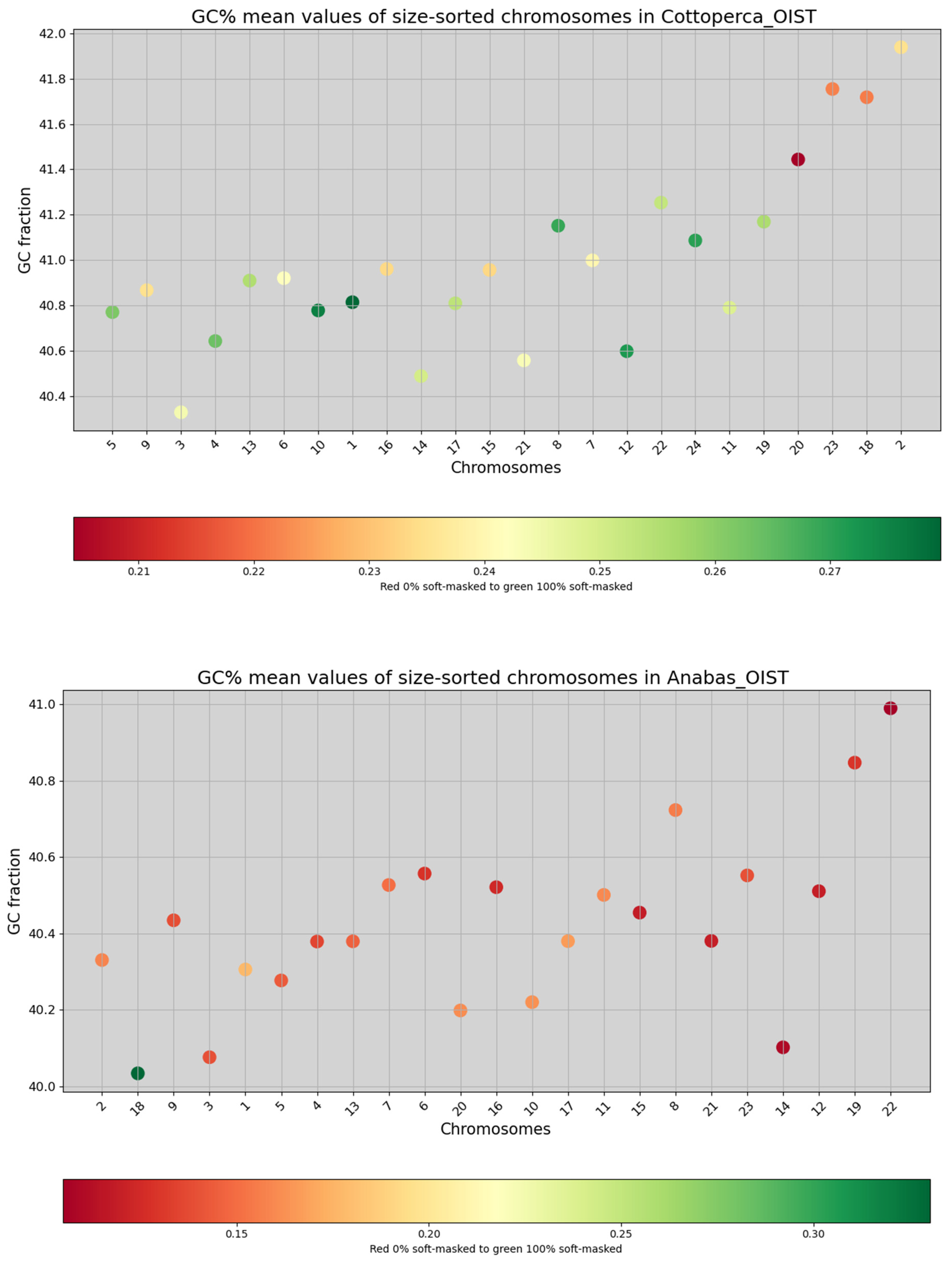
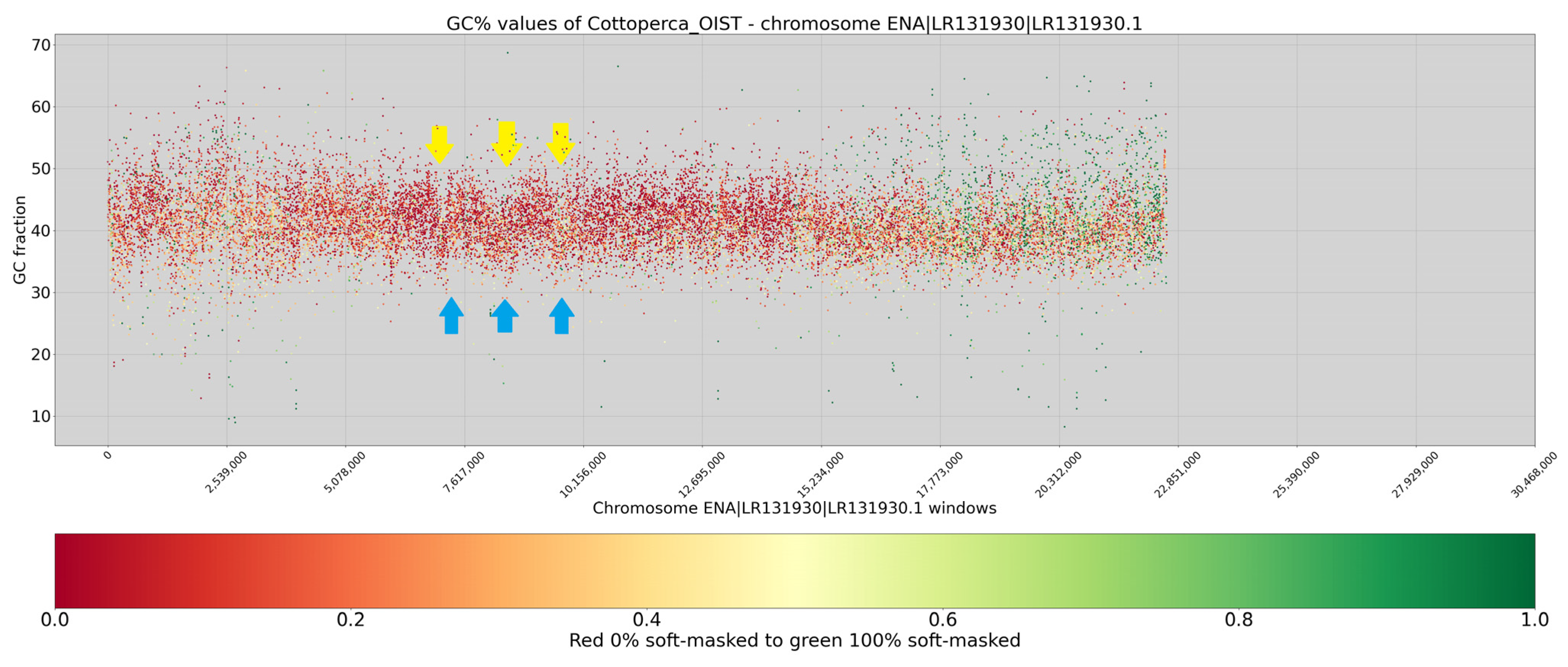
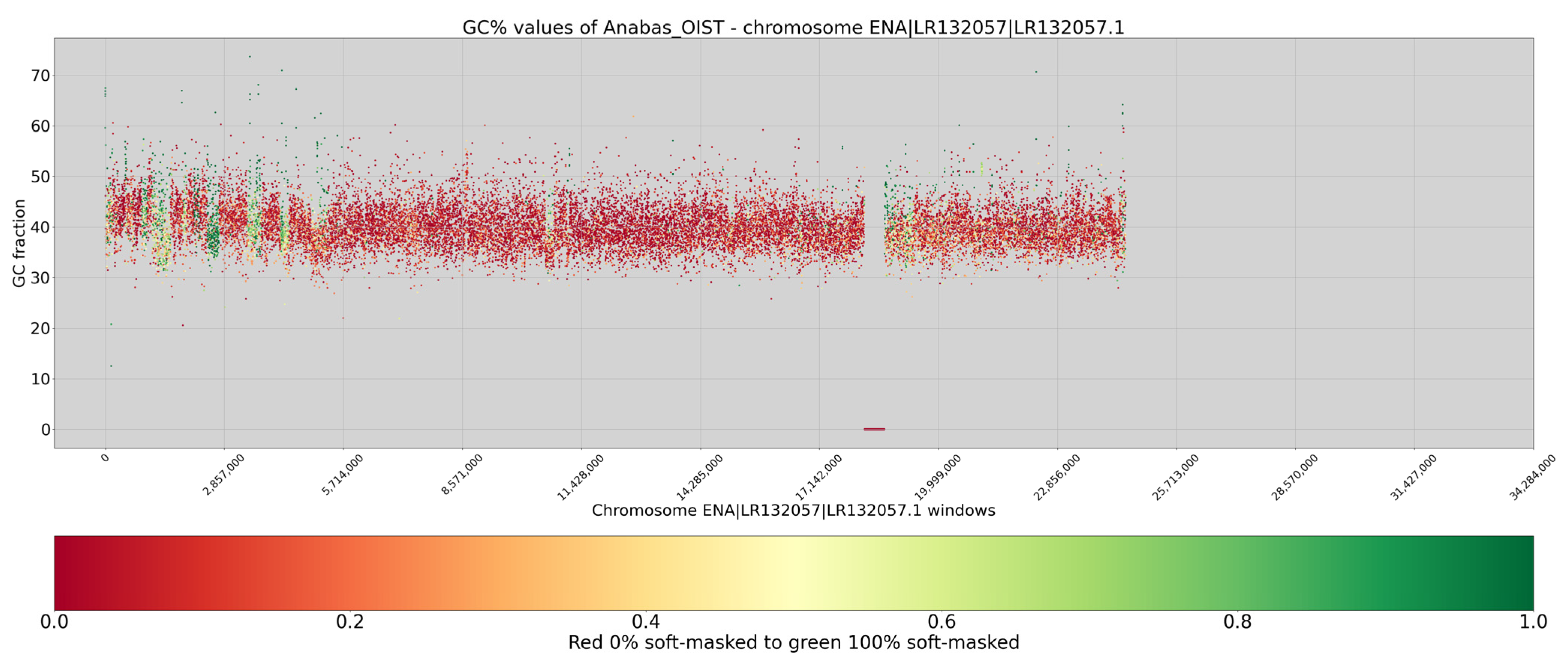
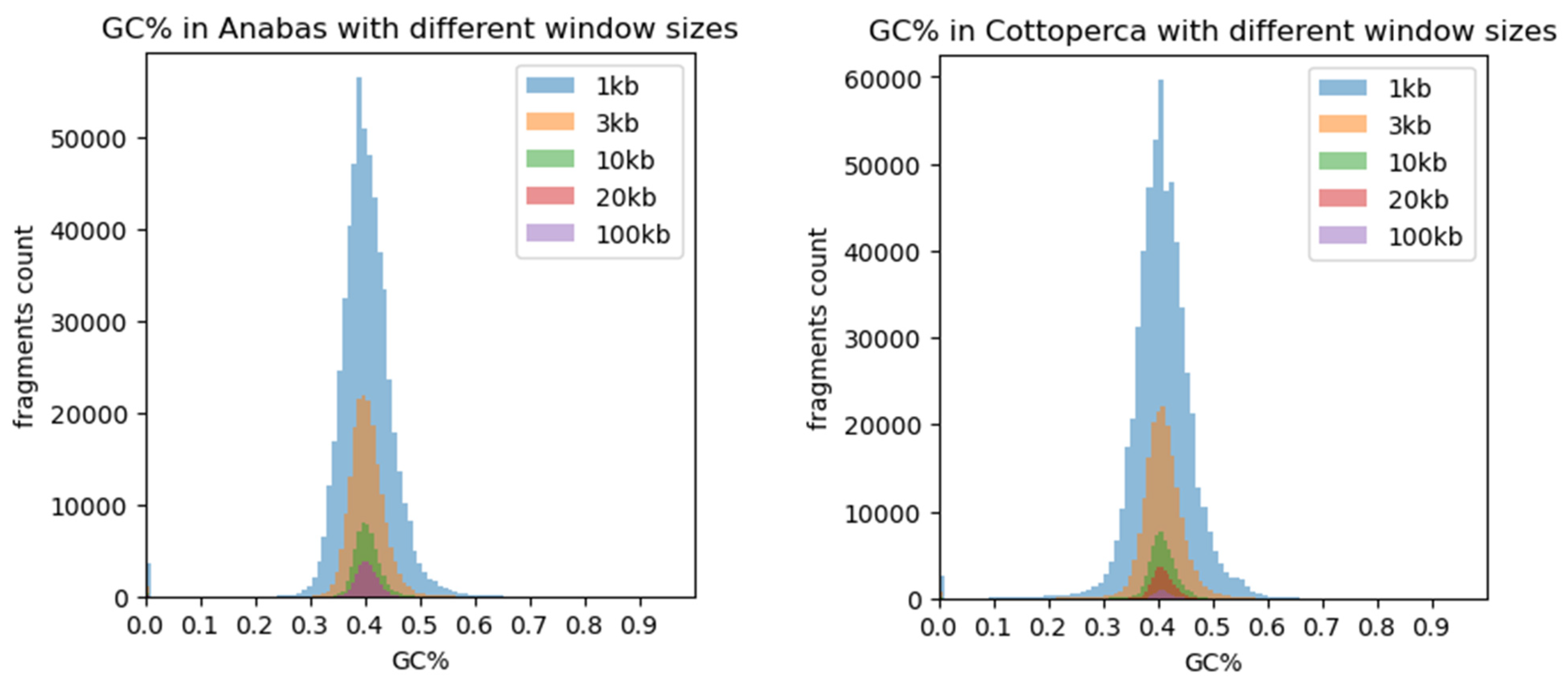
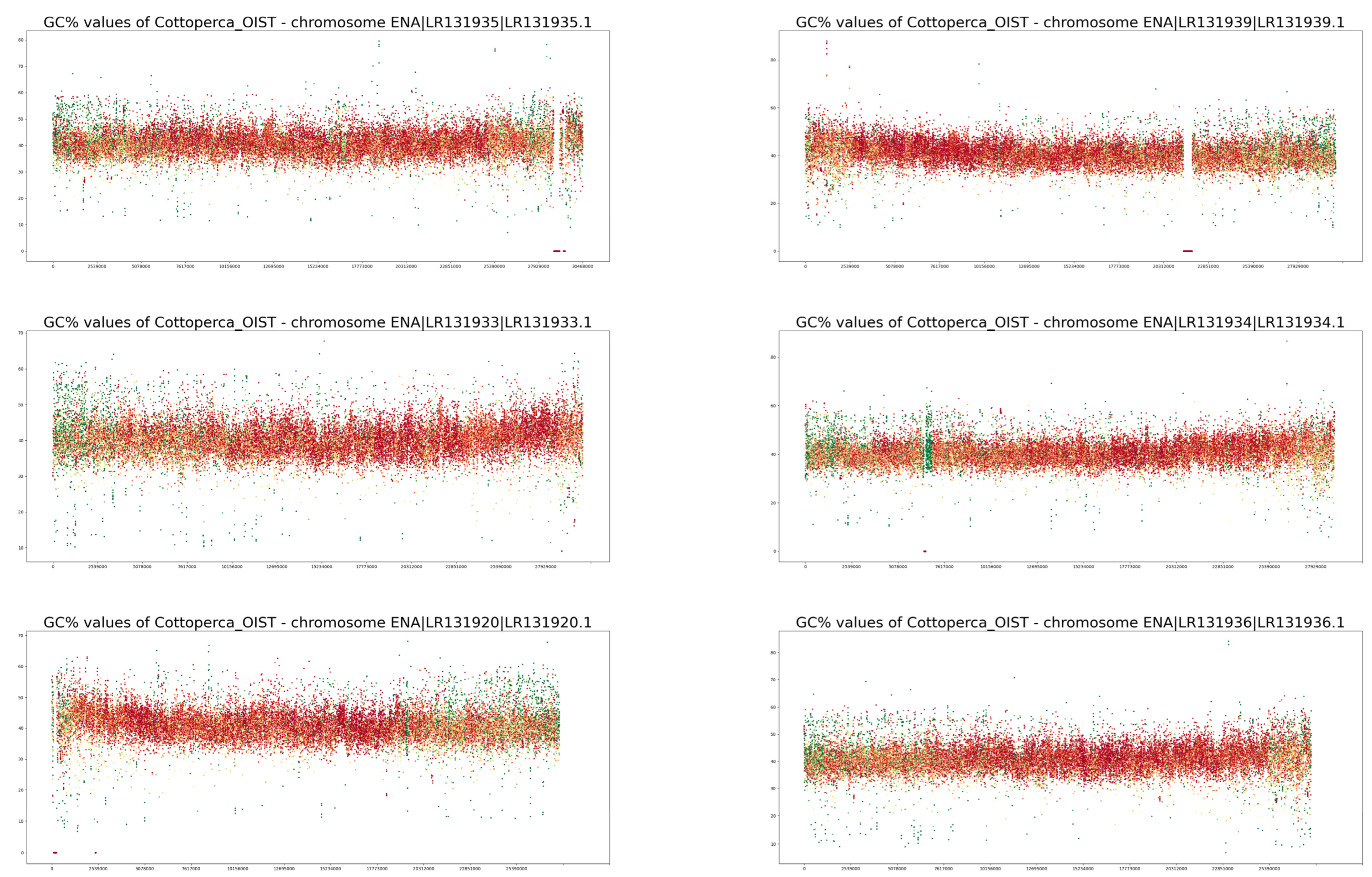
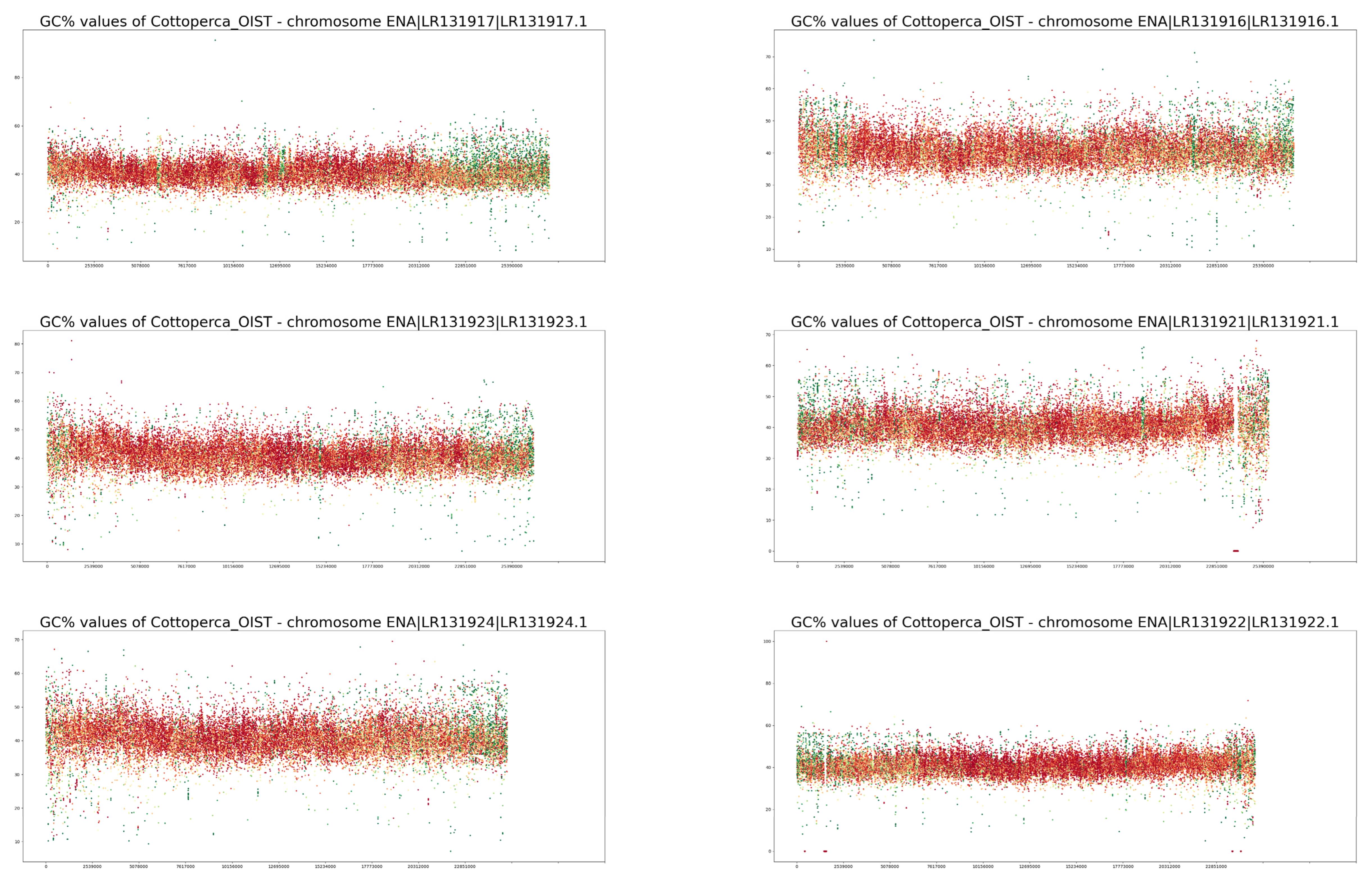
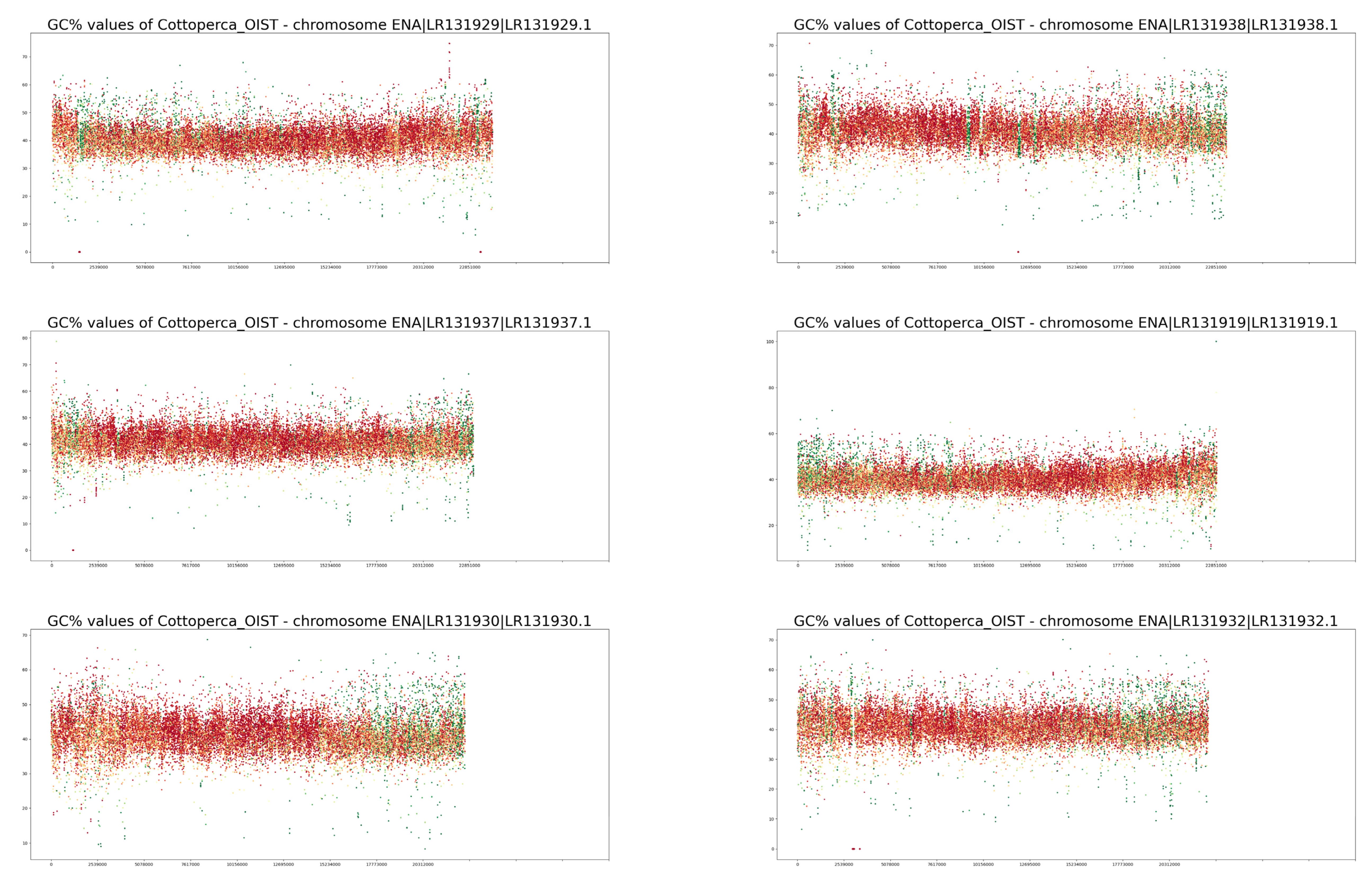
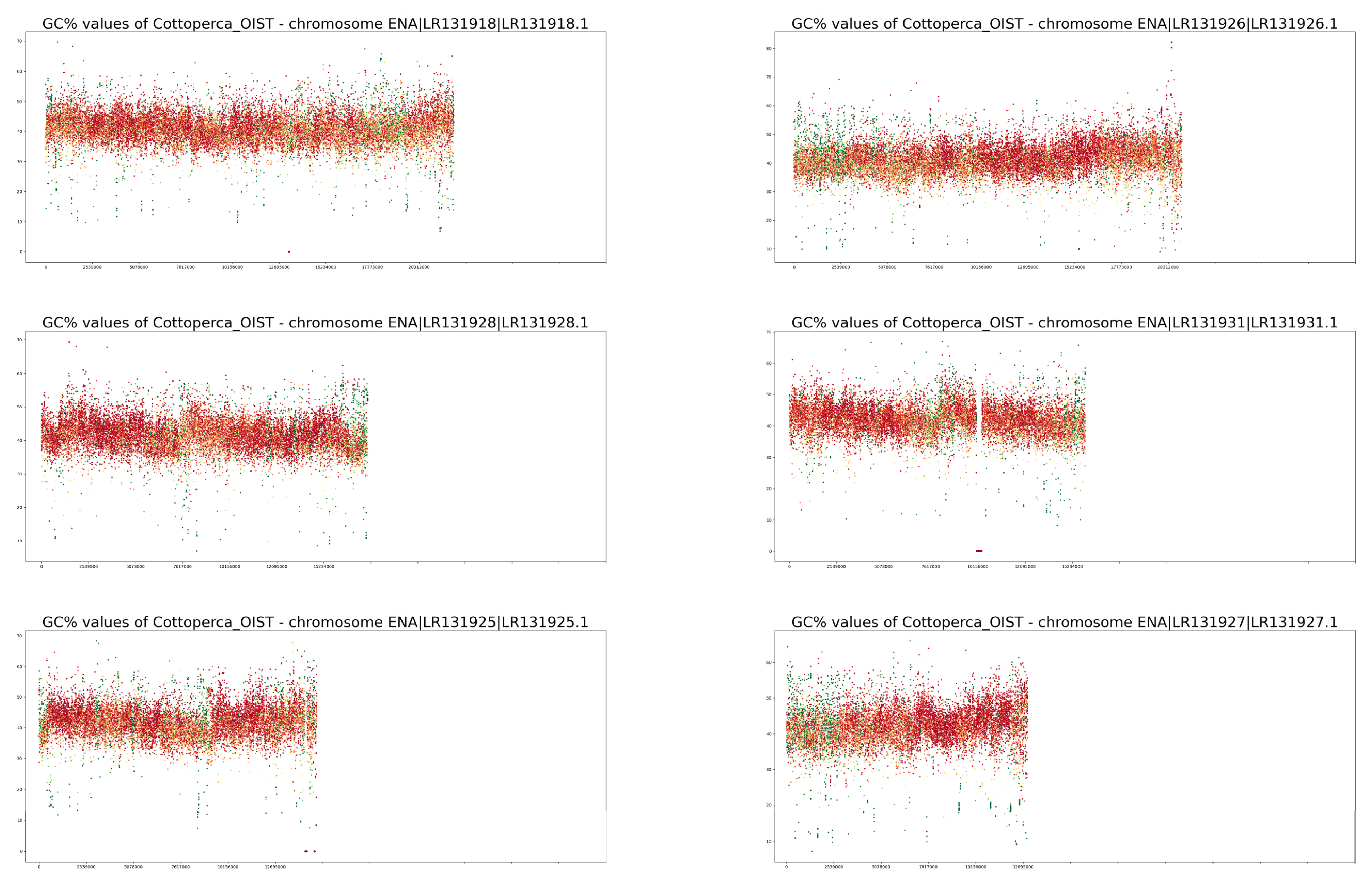
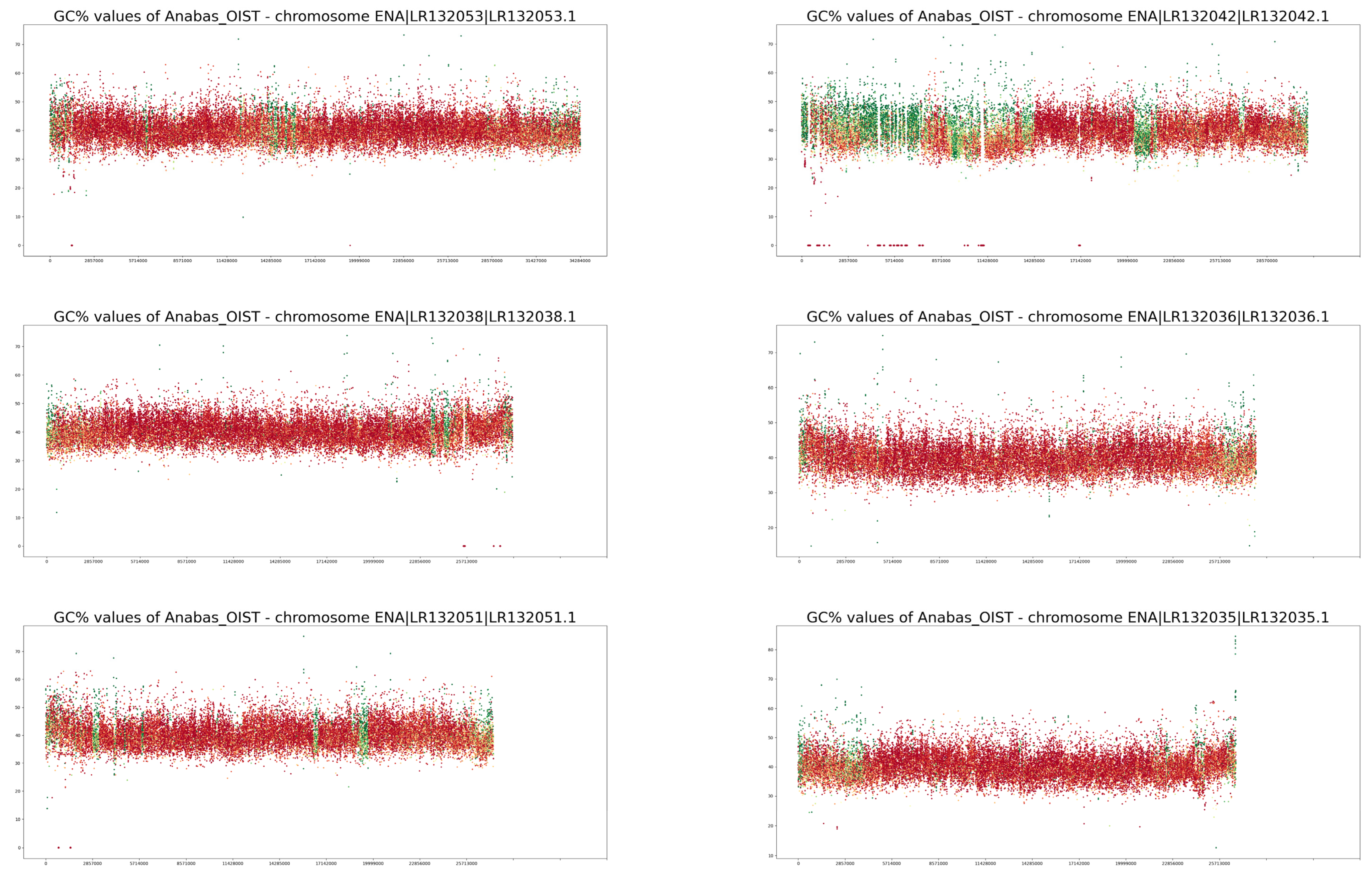
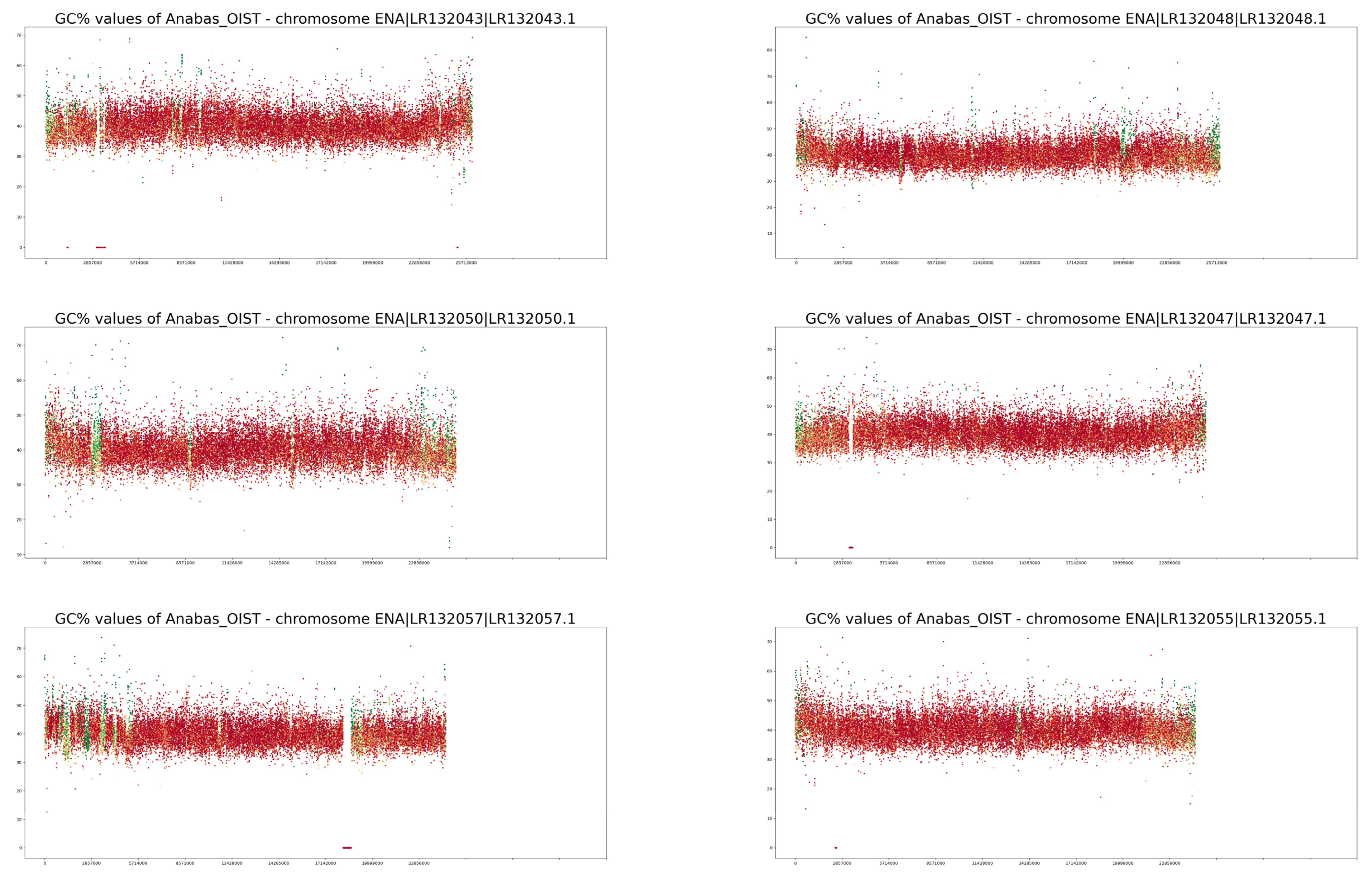
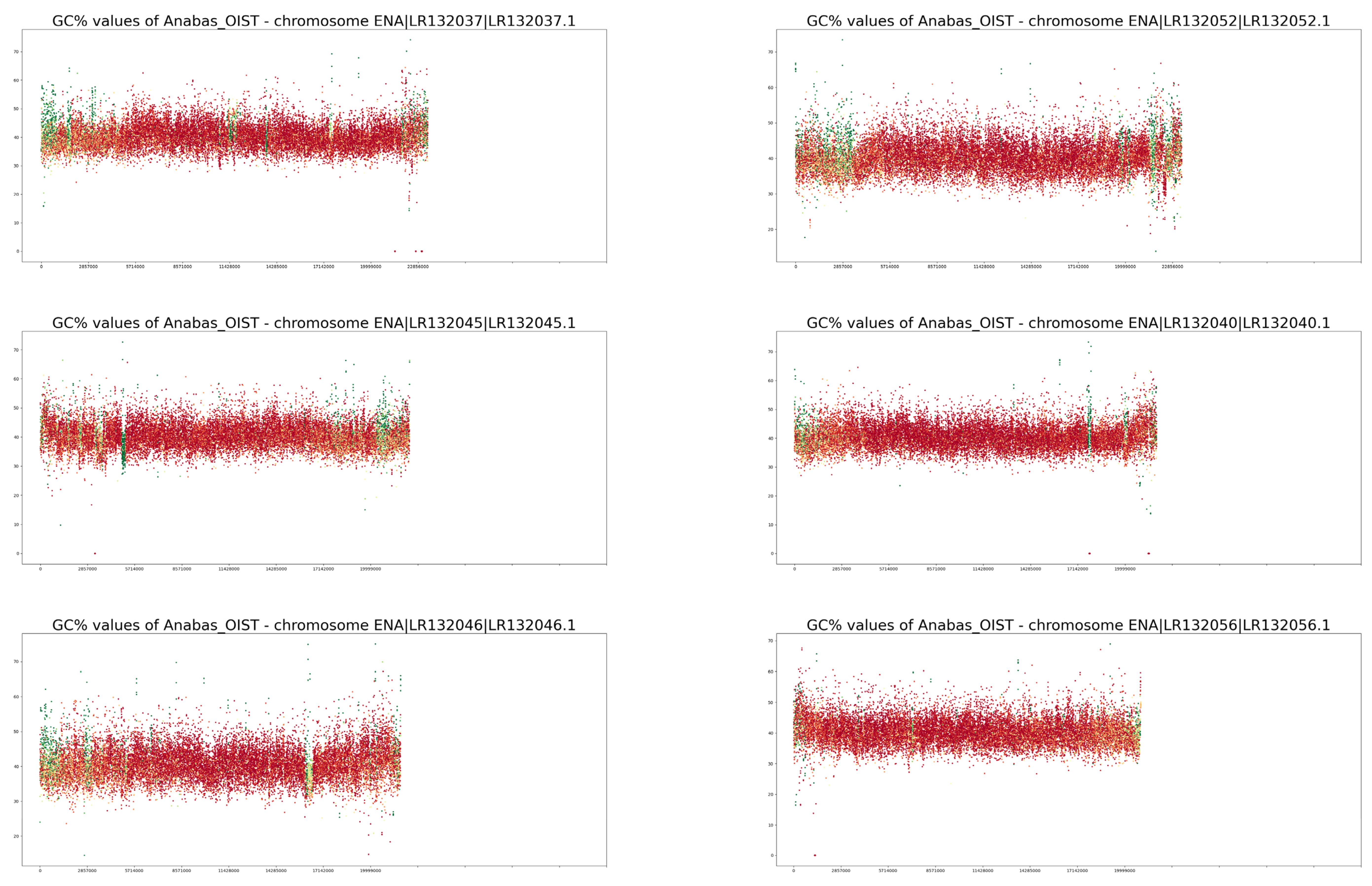
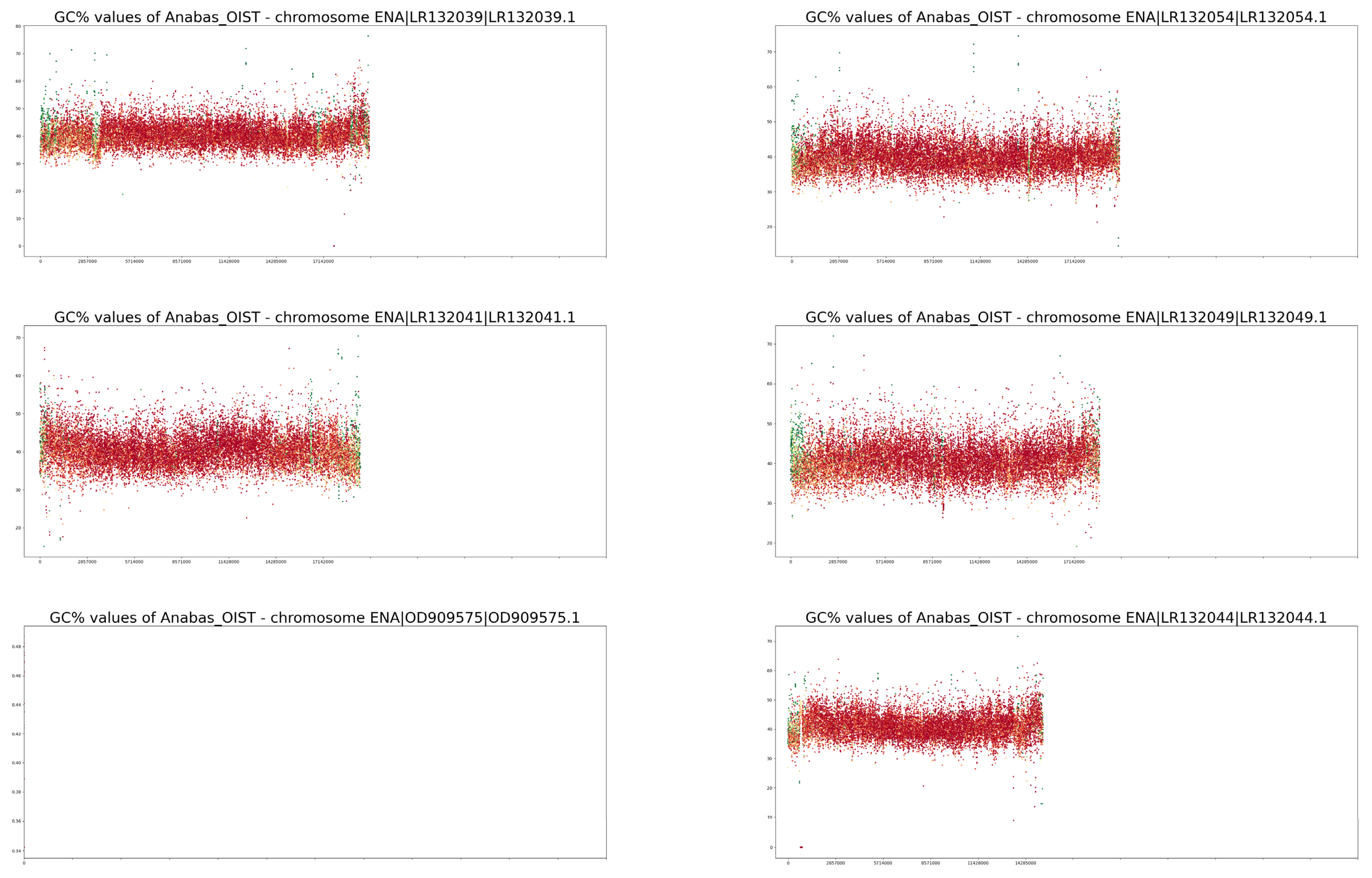
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








References
- Matoulek, D.; Borůvková, V.; Ocalewicz, K.; Symonová, R. GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics. Genes 2020, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Borůvková, V.; Howell, W.M.; Matoulek, D.; Symonová, R. Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution. Genes 2021, 12, 312. [Google Scholar] [CrossRef]
- Knytl, M.; Kalous, L.; Rab, P. Karyotype and Chromosome Banding of Endangered Crucian Carp, Carassius Carassius (Linnaeus, 1758) (Teleostei, Cyprinidae). CCG 2013, 7, 205–213. [Google Scholar] [CrossRef]
- Knytl, M.; Kalous, L.; Symonová, R.; Rylková, K.; Ráb, P. Chromosome Studies of European Cyprinid Fishes: Cross-Species Painting Reveals Natural Allotetraploid Origin of a Carassius Female with 206 Chromosomes. Cytogenet. Genome Res. 2013, 139, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Knytl, M.; Fornaini, N. Measurement of Chromosomal Arms and FISH Reveal Complex Genome Architecture and Standardized Karyotype of Model Fish, Genus Carassius. Cells 2021, 10, 2343. [Google Scholar] [CrossRef]
- Bertollo, L.A.C.; Fontes, M.S.; Fenocchio, A.S.; Cano, J. The X1X2Y Sex Chromosome System in the Fish Hoplias Malabaricus. I. G-, C- and Chromosome Replication Banding. Chromosome Res. 1997, 5, 493–499. [Google Scholar] [CrossRef]
- Gold, J.R.; Li, Y.C. Trypsin G-Banding of North American Cyprinid Chromosomes: Phylogenetic Considerations, Implications for Fish Chromosome Structure, and Chromosomal Polymorphism. Cytologia 1991, 56, 199–208. [Google Scholar] [CrossRef]
- Medrano, L.; Bernardi, G.; Couturier, J.; Dutrillaux, B.; Bernardi, G. Chromosome Banding and Genome Compartmentalization in Fishes. Chromosoma 1988, 96, 178–183. [Google Scholar] [CrossRef]
- Wiberg, U.H. Sex Determination in the European Eel (Anguilla Anguilla, L.). Cytogenet. Genome Res. 1983, 36, 589–598. [Google Scholar] [CrossRef]
- Luo, C. Multiple Chromosomal Banding in Grass Carp, Ctenopharyngodon Idellus. Heredity 1998, 81, 481–485. [Google Scholar] [CrossRef]
- Swarça, A.C.; Fenocchio, A.S.; Cestari, M.M.; Dias, A.L. First Chromosome Data on Steindachneridion Scripta (Pisces, Siluriformes, Pimelodidae) from Brazilian Rivers: Giemsa, CBG, G-, and RE Banding. Genet. Mol. Res. 2005, 4, 734–741. [Google Scholar] [PubMed]
- Jankun, M.; Mochol, M.; Ocalewicz, K. Conventional and Molecular Cytogenetics of the Pikeperch (Sander Lucioperca L.). Aquac. Res. 2014, 45, 1084–1089. [Google Scholar] [CrossRef]
- Jankun, M.; Ocalewicz, K.; Mochol, M. Chromosome Banding Studies by Replication and Restriction Enzyme Treatment in Vendace (Coregonus Albula) (Salmonidae, Salmoniformes). Folia Biol Krakow 2004, 52, 47–51. [Google Scholar]
- Jankun, M.; Ocalewicz, K.; Woznicki, P. Replication, C- and Fluorescent Chromosome Banding Patterns in European Whitefish, Coregonus Lavavetus L. Hereditas 2004, 128, 195–199. [Google Scholar] [CrossRef]
- de Araújo, W.C.; Martínez, P.A.; Molina, W.F. Mapping of Ribosomal DNA by FISH, EcoRI Digestion and Replication Bands in the Cardinalfish Apogon Americanus (Perciformes). Cytologia 2010, 75, 109–117. [Google Scholar] [CrossRef]
- Viñas, A.; Gómez, C.; Martínez, P.; Sánchez, L. Induction of G-Bands on Anguilla Anguilla Chromosomes by the Restriction Endonucleases HaeLll, HinfI, and MseI. Cytogenet. Genome Res. 1994, 65, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Symonová, R.; Havelka, M.; Amemiya, C.T.; Howell, W.M.; Kořínková, T.; Flajšhans, M.; Gela, D.; Ráb, P. Molecular Cytogenetic Differentiation of Paralogs of Hox Paralogs in Duplicated and Re-Diploidized Genome of the North American Paddlefish (Polyodon Spathula). BMC Genet. 2017, 18, 19. [Google Scholar] [CrossRef]
- Symonová, R.; Majtánová, Z.; Arias-Rodriguez, L.; Mořkovský, L.; Kořínková, T.; Cavin, L.; Pokorná, M.J.; Doležálková, M.; Flajšhans, M.; Normandeau, E.; et al. Genome Compositional Organization in Gars Shows More Similarities to Mammals than to Other Ray-Finned Fish. J. Exp. Zool. (Mol. Dev. Evol.) 2017, 328, 607–619. [Google Scholar] [CrossRef]
- Majtánová, Z.; Symonová, R.; Arias-Rodriguez, L.; Sallan, L.; Ráb, P. “Holostei versus Halecostomi” Problem: Insight from Cytogenetics of Ancient Nonteleost Actinopterygian Fish, Bowfin Amia Calva. J. Exp. Zool. (Mol. Dev. Evol.) 2017, 328, 620–628. [Google Scholar] [CrossRef]
- Matoulek, D.; Ježek, B.; Vohnoutová, M.; Symonová, R. Advances in Vertebrate (Cyto)Genomics Shed New Light on Fish Compositional Genome Evolution. Genes 2023, 14, 244. [Google Scholar] [CrossRef]
- Bernardi, G. The Neoselectionist Theory of Genome Evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 8385–8390. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N. Fine-Scale Quantification of GC-Biased Gene Conversion Intensity in Mammals. Peer Community J. 2021, 1, e17. [Google Scholar] [CrossRef]
- Symonová, R.; Suh, A. Nucleotide Composition of Transposable Elements Likely Contributes to AT/GC Compositional Homogeneity of Teleost Fish Genomes. Mobile DNA 2019, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S. On the Base Composition of Transposable Elements. Int. J. Mol. Sci. 2022, 23, 4755. [Google Scholar] [CrossRef]
- Gaffaroglu, M.; Majtánová, Z.; Symonová, R.; Pelikánová, Š.; Unal, S.; Lajbner, Z.; Ráb, P. Present and Future Salmonid Cytogenetics. Genes 2020, 11, 1462. [Google Scholar] [CrossRef]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards Complete and Error-Free Genome Assemblies of All Vertebrate Species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- National Library of Medicine (US). National Center for Biotechnology Information (NCBI) [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 1998. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 28 March 2023).
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Cozzi, P.; Milanesi, L.; Bernardi, G. Segmenting the Human Genome into Isochores. Evol. Bioinform. 2015, 11, EBO-S27693. [Google Scholar] [CrossRef]
- Lamolle, G.; Protasio, A.V.; Iriarte, A.; Jara, E.; Simón, D.; Musto, H. An Isochore-Like Structure in the Genome of the Flatworm Schistosoma Mansoni. Genome Biol. Evol. 2016, 8, 2312–2318. [Google Scholar] [CrossRef]
- Jakt, L.M.; Dubin, A.; Johansen, S.D. Intron Size Minimisation in Teleosts. BMC Genom. 2022, 23, 628. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.P.; Joyce, D.A.; Humphries, S.; Tindall, K.J.; Lunt, D.H. Comparative Analysis of Teleost Genome Sequences Reveals an Ancient Intron Size Expansion in the Zebrafish Lineage. Genome Biol. Evol. 2011, 3, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, F.; Ghigliotti, L.; Lecointre, G.; Ozouf-Costaz, C.; Coutanceau, J.-P.; Detrich, W.; Pisano, E. Karyotypes of Basal Lineages in Notothenioid Fishes: The Genus Bovichtus. Polar Biol. 2006, 29, 1071. [Google Scholar] [CrossRef]
- Kabir, M.A.; Habib, M.A.; Hasan, M.; Alam, S.S. Genetic Diversity in Three Forms of Anabas Testudineus Bloch. Cytologia 2012, 77, 231–237. [Google Scholar] [CrossRef]
- Khuda-Bukhsh, A.R.; Chakrabarti, C. Differential C-Heterochromatin Distribution in Two Species of Freshwater Fish, Anabas Testudineus (Bloch.) and Puntius Sarana (Hamilton.). Indian J. Exp. Biol. 2000, 38, 265–268. [Google Scholar]
- Tinni, S.R.; Jessy, N.S.; Hasan, M.M.; Mustafa, M.G.; Alam, S.S. Comparative Karyotype Analysis with Differential Staining in Two Forms of Anabas Testudineus Bloch. Cytologia 2007, 72, 71–75. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Pisano, E.; Ozouf-Costaz, C.; Hureau, J.-C.; Williams, R. Chromosome Differentiation in the Subantarctic Bovichtidae Species Cottoperca Gobio (Günther, 1861) and Pseudaphritis Urvillii (Valenciennes, 1832) (Pisces, Perciformes). Antart. Sci. 1995, 7, 381–386. [Google Scholar] [CrossRef]
- Pisano, E.; Ozouf-Costaz, C. Chromosome Change and the Evolution in the Antarctic Fish Suborder Notothenioidei. Antart. Sci. 2000, 12, 334–342. [Google Scholar] [CrossRef]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenet. Genome Res. 2015, 147, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Liu, S.; Zhou, T.; Tian, C.; Bao, L.; Dunham, R.; Liu, Z. Comparative Genome Analysis of 52 Fish Species Suggests Differential Associations of Repetitive Elements with Their Living Aquatic Environments. BMC Genom. 2018, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic Salmon Genome Provides Insights into Rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, X.; Xiao, Y.; Zhao, H.; Xu, S.; Wang, Y.; Wu, L.; Zhou, L.; Du, T.; Lv, X.; et al. Sequencing of the Black Rockfish Chromosomal Genome Provides Insight into Sperm Storage in the Female Ovary. DNA Res. 2019, 26, 453–464. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De Novo Assembly of the Aedes Aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait/Species | Anabas testudineus | Cottoperca gobio |
|---|---|---|
| Order | Anabantiformes | Perciformes |
| Family | Anabantidae | Bovichtidae |
| Diploid chromosome number (2n) | 46 | 48 |
| Genome assembly size (Mb) | 555.6 | 609.4 |
| NCBI GC% | 40.46% | 41% |
| GC% calculated in this study | 40.40% | 40.96% |
| Proportion of soft-masked regions (orig.) 1 | 11.52% | 18.15% |
| Proportion of soft-masked regions (new) 2 | 15.2% | 25.01% |
| GC% of soft-masked/repetitive regions | 38.40% | 40.54% |
| GC% of unmasked/non-repetitive regions | 40.66% | 41.05% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vohnoutová, M.; Žifčáková, L.; Symonová, R. Hidden Compositional Heterogeneity of Fish Chromosomes in the Era of Polished Genome Assemblies. Fishes 2023, 8, 185. https://doi.org/10.3390/fishes8040185
Vohnoutová M, Žifčáková L, Symonová R. Hidden Compositional Heterogeneity of Fish Chromosomes in the Era of Polished Genome Assemblies. Fishes. 2023; 8(4):185. https://doi.org/10.3390/fishes8040185
Chicago/Turabian StyleVohnoutová, Marta, Lucia Žifčáková, and Radka Symonová. 2023. "Hidden Compositional Heterogeneity of Fish Chromosomes in the Era of Polished Genome Assemblies" Fishes 8, no. 4: 185. https://doi.org/10.3390/fishes8040185
APA StyleVohnoutová, M., Žifčáková, L., & Symonová, R. (2023). Hidden Compositional Heterogeneity of Fish Chromosomes in the Era of Polished Genome Assemblies. Fishes, 8(4), 185. https://doi.org/10.3390/fishes8040185