
Siamese Fighting Fish (Betta splendens Regan) Gut Microbiota Associated with Age and Gender

 , , and
, , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Betta Fish Sampling and Extraction of Microbial DNA and Measurements of Physiological Parameters
2.2. Extraction of Microbial Genomic DNA
2.3. 16S rRNA Gene Amplicon Sequencing
2.4. Bioinformatics and Statistical Analysis
2.4.1. Taxonomic Annotation of Amplicon Sequence Variants (ASVs)
2.4.2. Alpha and Beta Diversity Analyses
2.4.3. Network Analysis
2.4.4. Functional Prediction of Gut Microbiota
3. Results
3.1. Microbial Diversity of the Siamese Fighting Fish Intestinal Tract
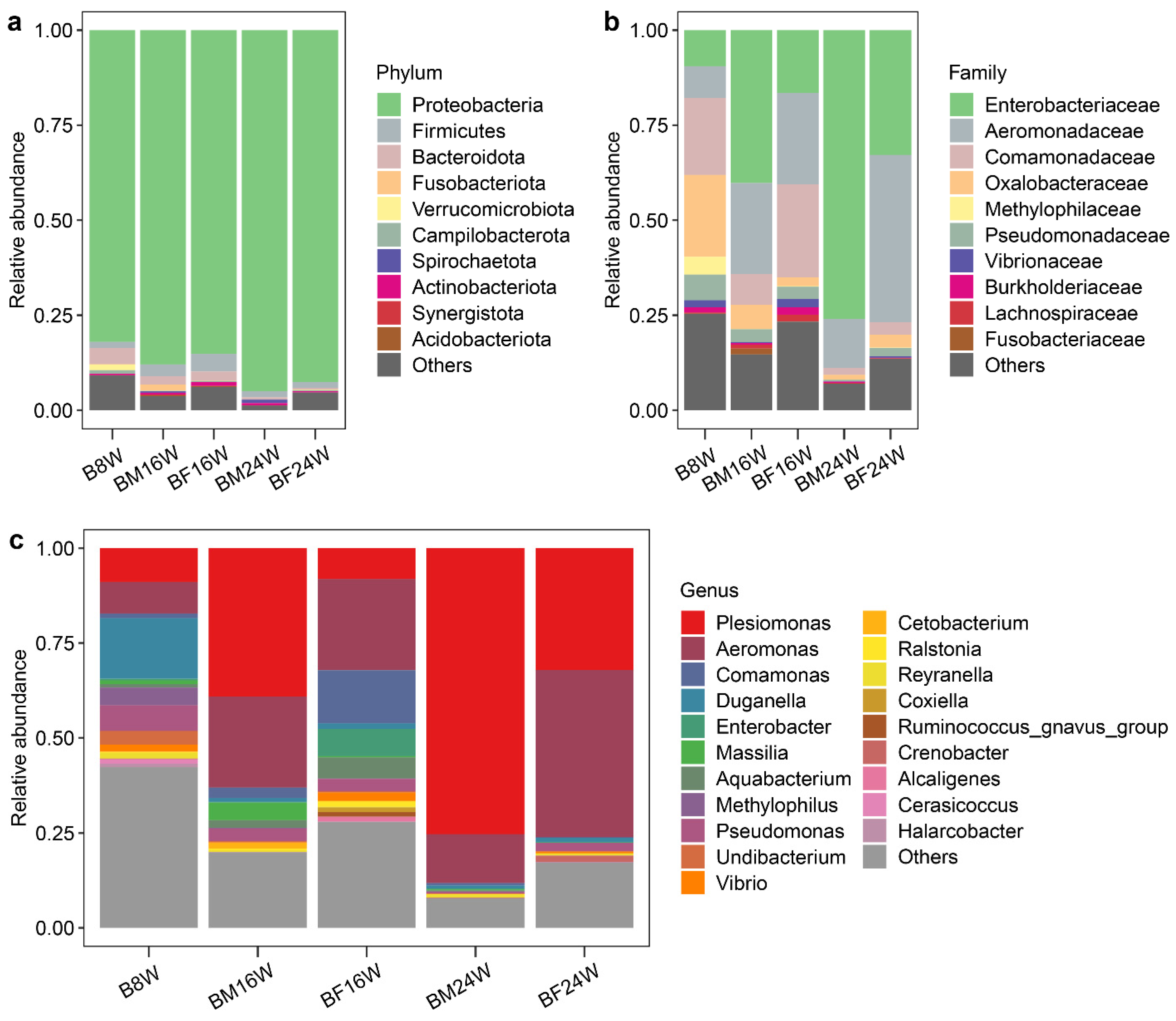
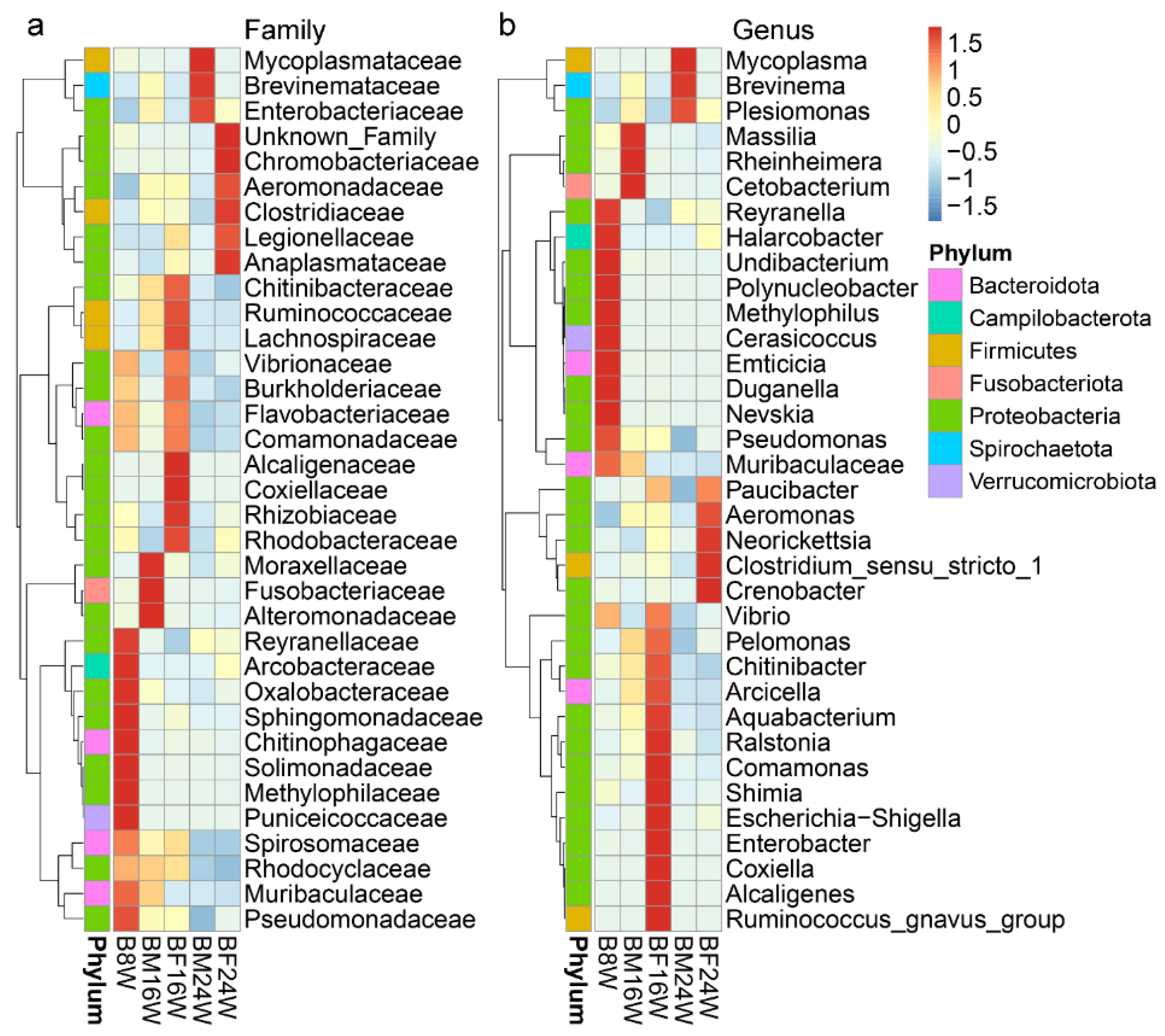
3.2. The Composition of Gut Microbiota in Young and Adult Siamese Fighting Fish
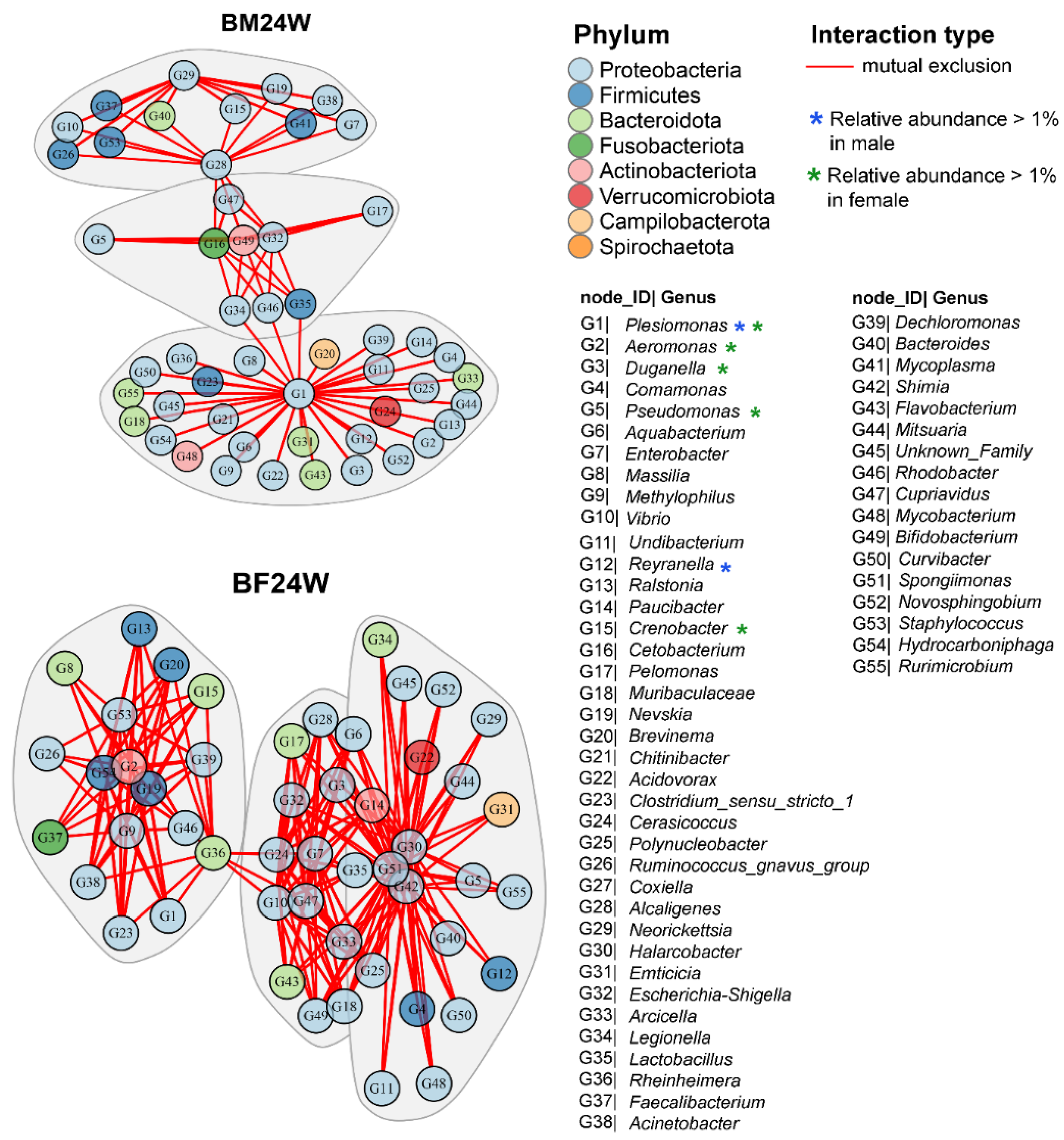
3.3. Gender Had a Significant Influence on the Structure of the Microbial Community Network of Siamese Fighting Fish
3.4. Associations between Siamese Fighting Fish Gut Microbiota and Predicted Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monvises, A.; Nuangsaeng, B.; Sriwattanarothai, N.; Panijpan, B. The Siamese fighting fish: Well-known generally but little-known scientifically. ScienceAsia 2009, 35, 8. [Google Scholar] [CrossRef]
- Monticini, P. The Ornamental Fish Trade (Production and Commerce of Ornamental Fish: Technical-Managerial and Legislative Aspects); Globefish Research Programme; Food and Agriculture Organization of the United Nations: Rome, Italy, 2010; Volume 102. [Google Scholar]
- Siamese Fighting Fish Sees Brighter Future. Available online: https://www.ditpthinkthailand.com/siamese-fighting-fish-sees-brighter-future/ (accessed on 4 September 2022).
- Betta Fish. Available online: https://www.nationalgeographic.com/animals/fish/facts/betta-fish (accessed on 5 September 2022).
- Rawls, J.F.; Samuel, B.S.; Gordon, J.I. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. USA 2004, 101, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Navarrete, P. 16S rDNA-Based analysis of dominant bacterial populations associated with early life stages of coho salmon (Oncorhynchus kisutch). Microb. Ecol. 2006, 51, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Ray, A.K. Bacterial symbiosis in the fish gut and its role in health and metabolism. Symbiosis 2017, 72, 1–11. [Google Scholar] [CrossRef]
- Ringø, E.; Strøm, E.; Tabachek, J.-A. Intestinal microflora of salmonids: A review. Aquac. Res. 1995, 26, 773–789. [Google Scholar] [CrossRef]
- Gómez, G.D.; Balcázar, J.L. A review on the interactions between gut microbiota and innate immunity of fish: Table 1. FEMS Immunol. Med. Microbiol. 2008, 52, 145–154. [Google Scholar] [CrossRef]
- Butt, R.L.; Volkoff, H. Gut microbiota and energy homeostasis in fish. Front. Endocrinol. 2019, 10, 6–8. [Google Scholar] [CrossRef]
- Thongprajukaew, K. Biology of Siamese fighting fish (Betta splendens Regan, 1910). KKU Sci. J. 2013, 41, 1–15. [Google Scholar]
- Piazzon, M.C.; Naya-Català, F.; Simó-Mirabet, P.; Picard-Sánchez, A.; Roig, F.J.; Calduch-Giner, J.A.; Sitjà-Bobadilla, A.; Pérez-Sánchez, J. Sex, age, and bacteria: How the intestinal microbiota is modulated in a protandrous hermaphrodite fish. Front. Microbiol. 2019, 10, 2512. [Google Scholar] [CrossRef] [PubMed]
- Rosado, D.; Pérez-Losada, M.; Pereira, A.; Severino, R.; Xavier, R. Effects of aging on the skin and gill microbiota of farmed seabass and seabream. Anim. Microbiome 2021, 3, 10. [Google Scholar] [CrossRef]
- Stephens, W.Z.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Devi, P.A.; Padmavathy, P.; Aanand, S.; Aruljothi, K. Review on water quality parameters in freshwater cage fish culture. Int. J. Appl. Res. 2017, 3, 114–120. [Google Scholar]
- Yavuzcan Yildiz, H.; Robaina, L.; Pirhonen, J.; Mente, E.; Domínguez, D.; Parisi, G. Fish welfare in aquaponic systems: Its relation to water quality with an emphasis on feed and faeces—A review. Water 2017, 9, 13. [Google Scholar] [CrossRef]
- Betta Fish Anatomy—Plus Male and Female Differences. Available online: https://bettafish.org/betta-fish-anatomy/ (accessed on 17 November 2022).
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shao, D.; Zhou, J.; Gu, J.; Qin, J.; Chen, W.; Wei, W. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin. J. Cancer Res. 2020, 32, 755–767. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal 2006, 1695, 1–9. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Rahnavard, G.; Franzosa, E.; McIver, L.; Schwager, E.; Lloyd-Price, J.; Weingart, G.; Huttenhower, C. High-sensitivity pattern discovery in large, paired multi-omic datasets. Bioinformatics 2022, 38, i378–i385. [Google Scholar]
- Zhang, Z.; Li, D.; Refaey, M.M.; Xu, W.; Tang, R.; Li, L. Host age affects the development of southern catfish gut bacterial community divergent from that in the food and rearing water. Front. Microbiol. 2018, 9, 495. [Google Scholar] [CrossRef]
- Cantas, L.; Sørby, J.R.T.; Aleström, P.; Sørum, H. Culturable gut microbiota diversity in zebrafish. Zebrafish 2012, 9, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Willemsen, D.; Popkes, M.; Metge, F.; Gandiwa, E.; Reichard, M.; Valenzano, D.R. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. eLife 2017, 6, e27014. [Google Scholar] [CrossRef] [PubMed]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef]
- Song, C.; Wen, H.; Liu, G.; Ma, X.; Lv, G.; Wu, N.; Chen, J.; Xue, M.; Li, H.; Xu, P. Gut microbes reveal Pseudomonas medicates ingestion preference via protein utilization and cellular homeostasis under feed domestication in freshwater drum, Aplodinotus grunniens. Front. Microbiol. 2022, 13, 1831. [Google Scholar] [CrossRef]
- Talwar, C.; Nagar, S.; Lal, R.; Negi, R.K. Fish gut microbiome: Current approaches and future perspectives. Indian J. Microbiol. 2018, 58, 397–414. [Google Scholar] [CrossRef]
- Wei, Y.; Bu, J.; Long, H.; Zhang, X.; Cai, X.; Huang, A.; Ren, W.; Xie, Z. Community structure of protease-producing bacteria cultivated from aquaculture systems: Potential impact of a tropical environment. Front. Microbiol. 2021, 12, 638129. [Google Scholar] [CrossRef]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef]
- Xiong, J.-B.; Nie, L.; Chen, J. Current understanding on the roles of gut microbiota in fish disease and immunity. Zool. Res. 2019, 40, 70–76. [Google Scholar] [PubMed]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Novoslavskij, A.; Terentjeva, M.; Eizenberga, I.; Valciņa, O.; Bartkevičs, V.; Bērziņš, A. Major foodborne pathogens in fish and fish products: A review. Ann. Microbiol. 2016, 66, 1–15. [Google Scholar] [CrossRef]
- Kim, P.S.; Shin, N.-R.; Lee, J.-B.; Kim, M.-S.; Whon, T.W.; Hyun, D.-W.; Yun, J.-H.; Jung, M.-J.; Kim, J.Y.; Bae, J.-W. Host habitat is the major determinant of the gut microbiome of fish. Microbiome 2021, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.K. Role of gastrointestinal microbiota in fish. Aquac. Res. 2010, 41, 1553–1573. [Google Scholar] [CrossRef]
- Larsen, A.M.; Mohammed, H.H.; Arias, C.R. Characterization of the gut microbiota of three commercially valuable warmwater fish species. J. Appl. Microbiol. 2014, 116, 1396–1404. [Google Scholar] [CrossRef]
- Pemberton, J.M.; Kidd, S.P.; Schmidt, R. Secreted enzymes of Aeromonas. FEMS Microbiol. Lett. 2006, 152, 1–10. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, C.; Yang, G.; Gong, X.; Chen, X.; Xu, L.; Bao, B. Cellulase-producing bacteria of Aeromonas are dominant and indigenous in the gut of Ctenopharyngodon idellus (Valenciennes). Aquac. Res. 2011, 42, 499–505. [Google Scholar] [CrossRef]
- Li, T.; Long, M.; Ji, C.; Shen, Z.; Gatesoupe, F.-J.; Zhang, X.; Zhang, Q.; Zhang, L.; Zhao, Y.; Liu, X.; et al. Alterations of the gut microbiome of largemouth bronze gudgeon (Coreius guichenoti) suffering from furunculosis. Sci. Rep. 2016, 6, 30606. [Google Scholar] [CrossRef]
- Riera, J.L.; Baldo, L. Microbial co-occurrence networks of gut microbiota reveal community conservation and diet-associated shifts in cichlid fishes. Anim. Microbiome 2020, 2, 36. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Li, J.; Chen, W. The gut microbiome composition and degradation enzymes activity of black Amur bream (Megalobrama terminalis) in response to breeding migratory behavior. Ecol. Evol. 2021, 11, 5150–5163. [Google Scholar] [CrossRef] [PubMed]
- Sultana, S.; Khan, M.N.; Hossain, M.S.; Dai, J.; Rahman, M.S.; Salimullah, M. Community structure and functional annotations of the skin microbiome in healthy and diseased catfish, Heteropneustes fossilis. Front. Microbiol. 2022, 13, 551. [Google Scholar] [CrossRef] [PubMed]
- Duman, M.; Mulet, M.; Altun, S.; Saticioglu, I.B.; Ozdemir, B.; Ajmi, N.; Lalucat, J.; García-Valdés, E. The diversity of Pseudomonas species isolated from fish farms in Turkey. Aquaculture 2021, 535, 736369. [Google Scholar] [CrossRef]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef]
- Wu, L.; Luo, Y. Bacterial quorum-sensing systems and their role in intestinal bacteria-host crosstalk. Front. Microbiol. 2021, 12, 611413. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement | Young | Female | Male | p-Value | ||
|---|---|---|---|---|---|---|
| B8W | BF16W | BF24W | BM16W | BM24W | ||
| Width (cm) | 0.22 ± 0.03 a | 0.81 ± 0.06 a,b | 0.69 ± 0.05 a,b | 0.86 ± 0.04 b | 0.85 ± 0.00 a,b | 0.02 k |
| Length (cm) | 0.87 ± 0.06 a | 3.29 ± 0.21 a,b | 3.19 ± 0.10 a,b | 3.82 ± 0.12 b | 3.61 ± 0.05 a,b | 0.01 k |
| Weight (g) | 0.09 ± 0.01 d | 0.39 ± 0.07 b,c | 0.31 ± 0.04 c | 0.63 ± 0.10 a | 0.50 ± 0.02 a,b | <0.0001 w |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruneck, L.; Jinatham, V.; Therdtatha, P.; Popluechai, S. Siamese Fighting Fish (Betta splendens Regan) Gut Microbiota Associated with Age and Gender. Fishes 2022, 7, 347. https://doi.org/10.3390/fishes7060347
Gruneck L, Jinatham V, Therdtatha P, Popluechai S. Siamese Fighting Fish (Betta splendens Regan) Gut Microbiota Associated with Age and Gender. Fishes. 2022; 7(6):347. https://doi.org/10.3390/fishes7060347
Chicago/Turabian StyleGruneck, Lucsame, Vasana Jinatham, Phatthanaphong Therdtatha, and Siam Popluechai. 2022. "Siamese Fighting Fish (Betta splendens Regan) Gut Microbiota Associated with Age and Gender" Fishes 7, no. 6: 347. https://doi.org/10.3390/fishes7060347
APA StyleGruneck, L., Jinatham, V., Therdtatha, P., & Popluechai, S. (2022). Siamese Fighting Fish (Betta splendens Regan) Gut Microbiota Associated with Age and Gender. Fishes, 7(6), 347. https://doi.org/10.3390/fishes7060347