
Comparative Transcriptome Analysis of Female and Male Fine-Patterned Puffer: Identification of Candidate Genes Associated with Growth and Sex Differentiation

Abstract
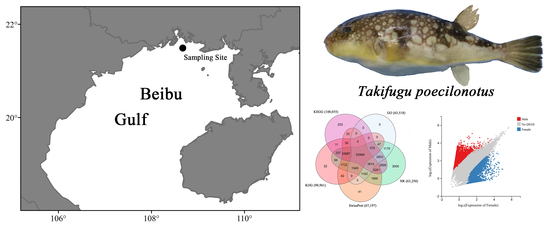
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. RNA Extraction, cDNA Library Building, and Transcriptome Sequencing
2.4. Assembly and Annotation
2.5. Analysis of Potential Candidate Genes
2.6. Simple Sequence Repeat (SSR) Loci Detection
2.7. Quantitative Real-Time PCR Validation
3. Results
3.1. Sequencing and Assembly
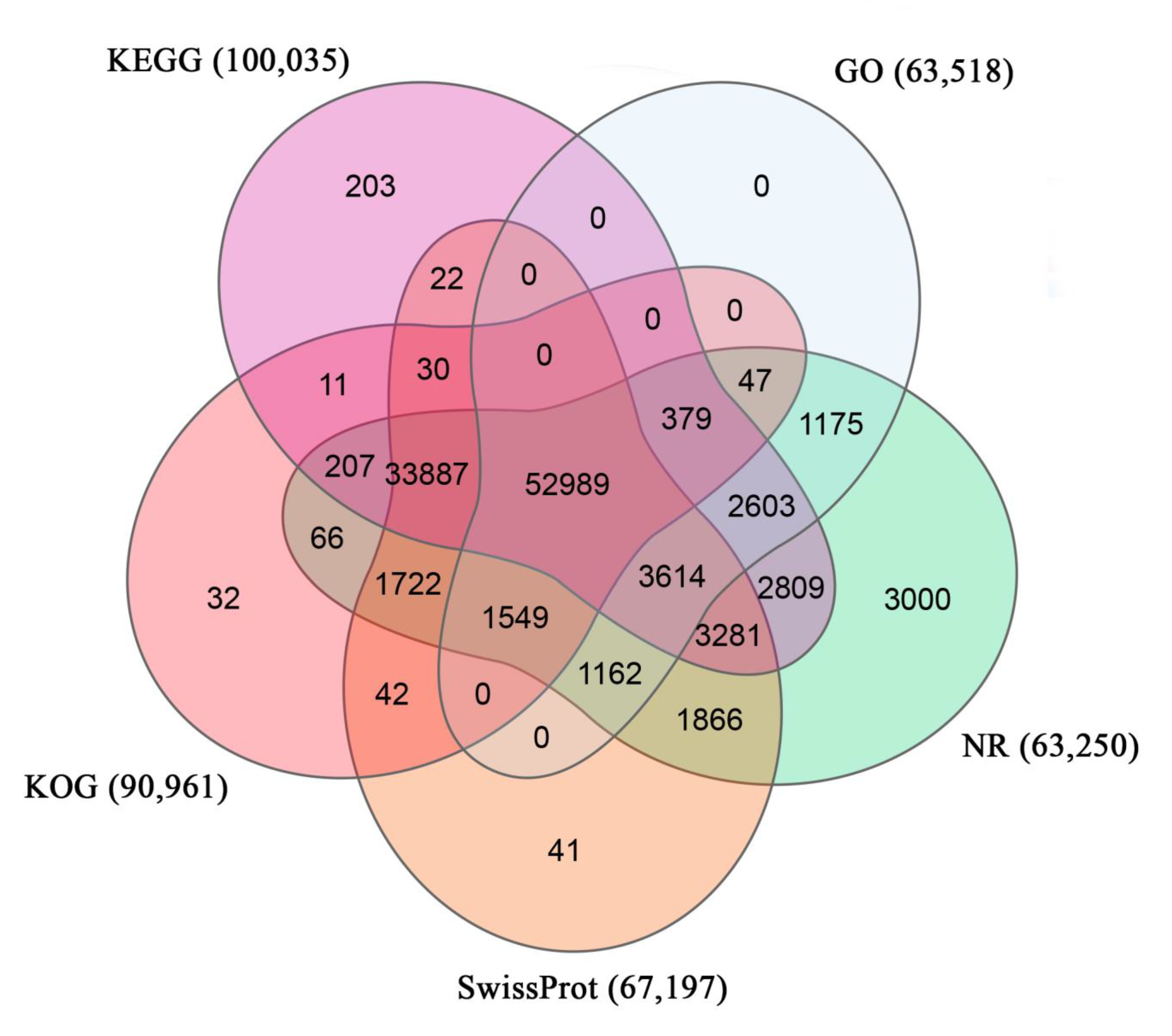
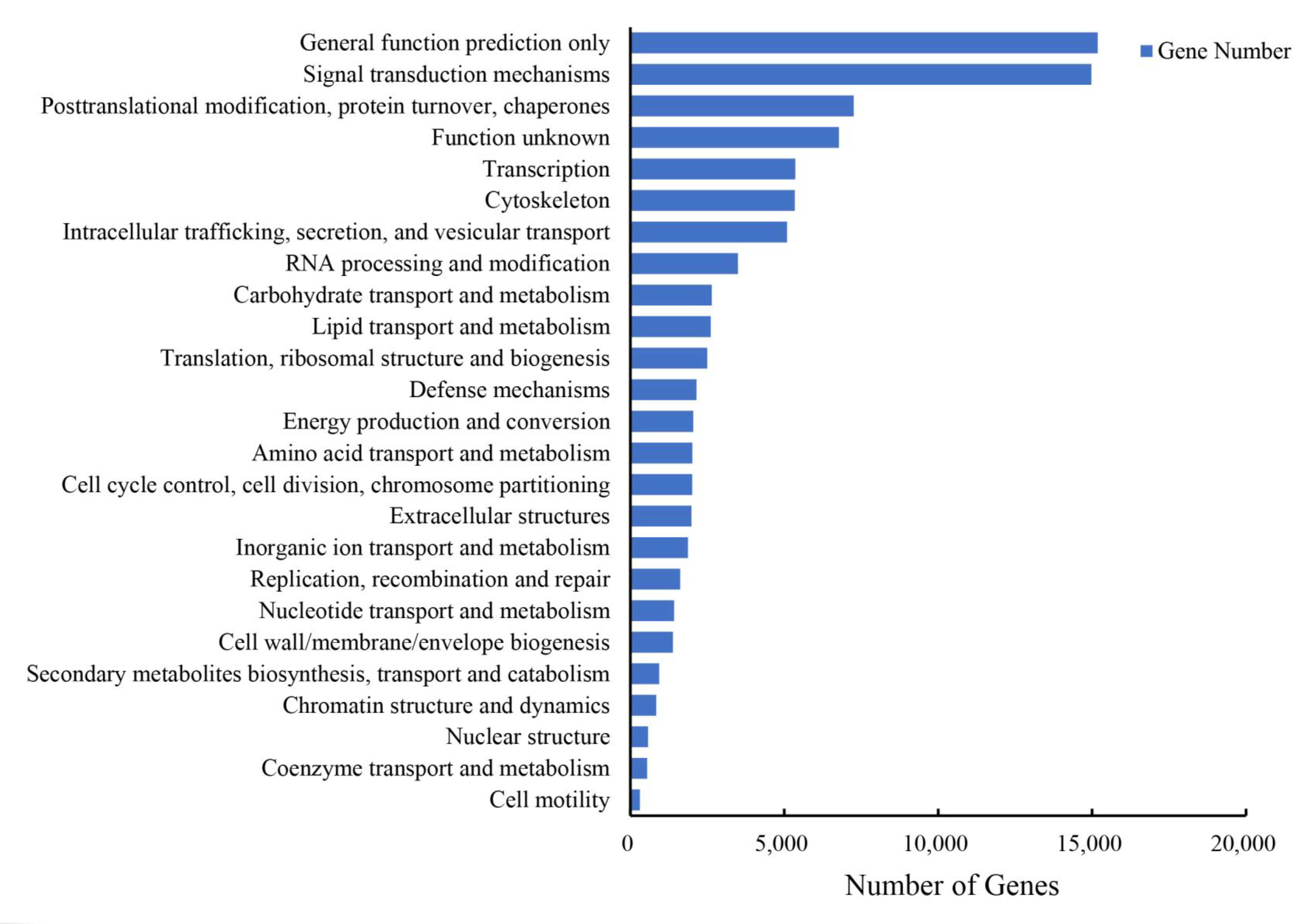
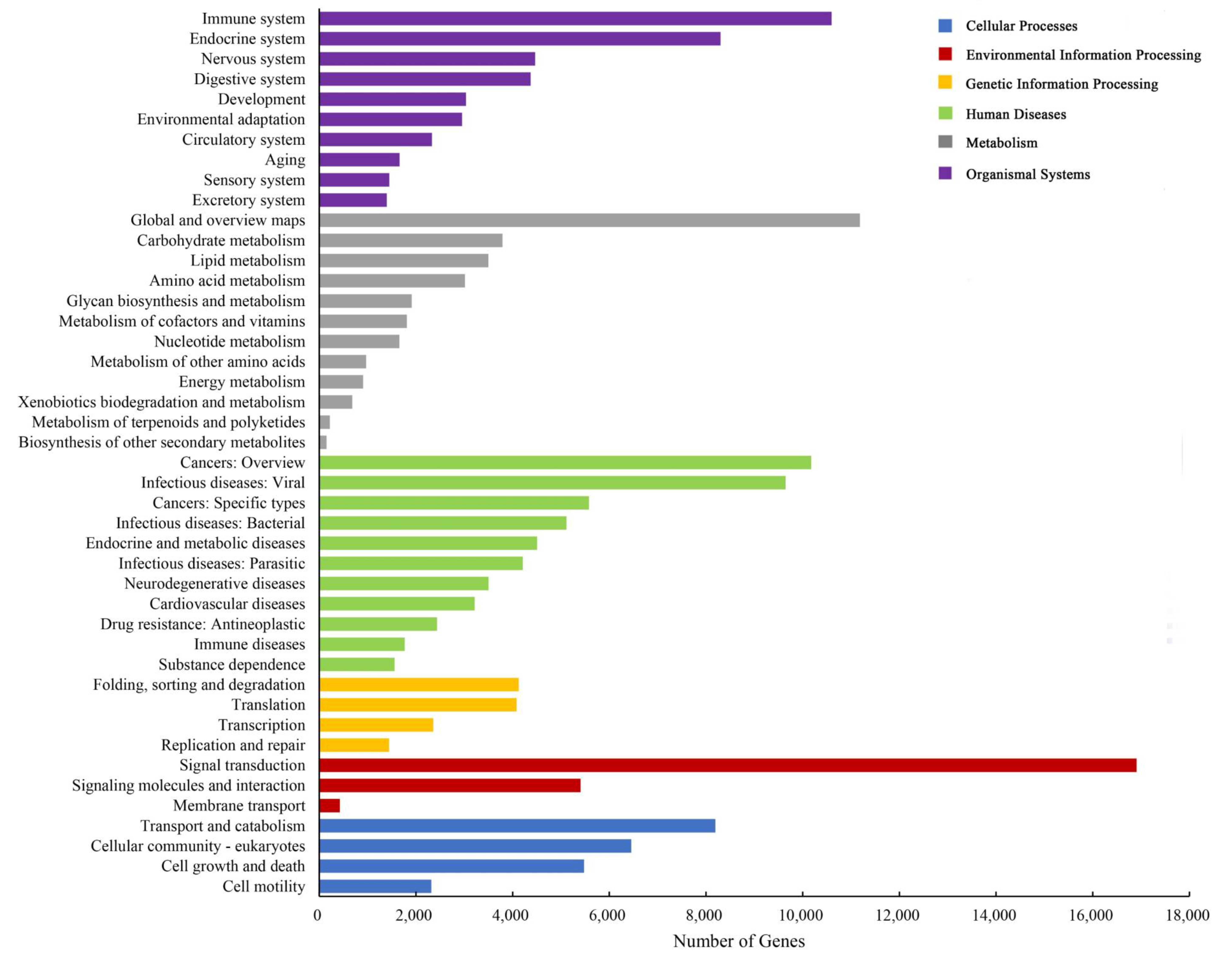
3.2. Annotation of T. poecilonotus Transcriptome
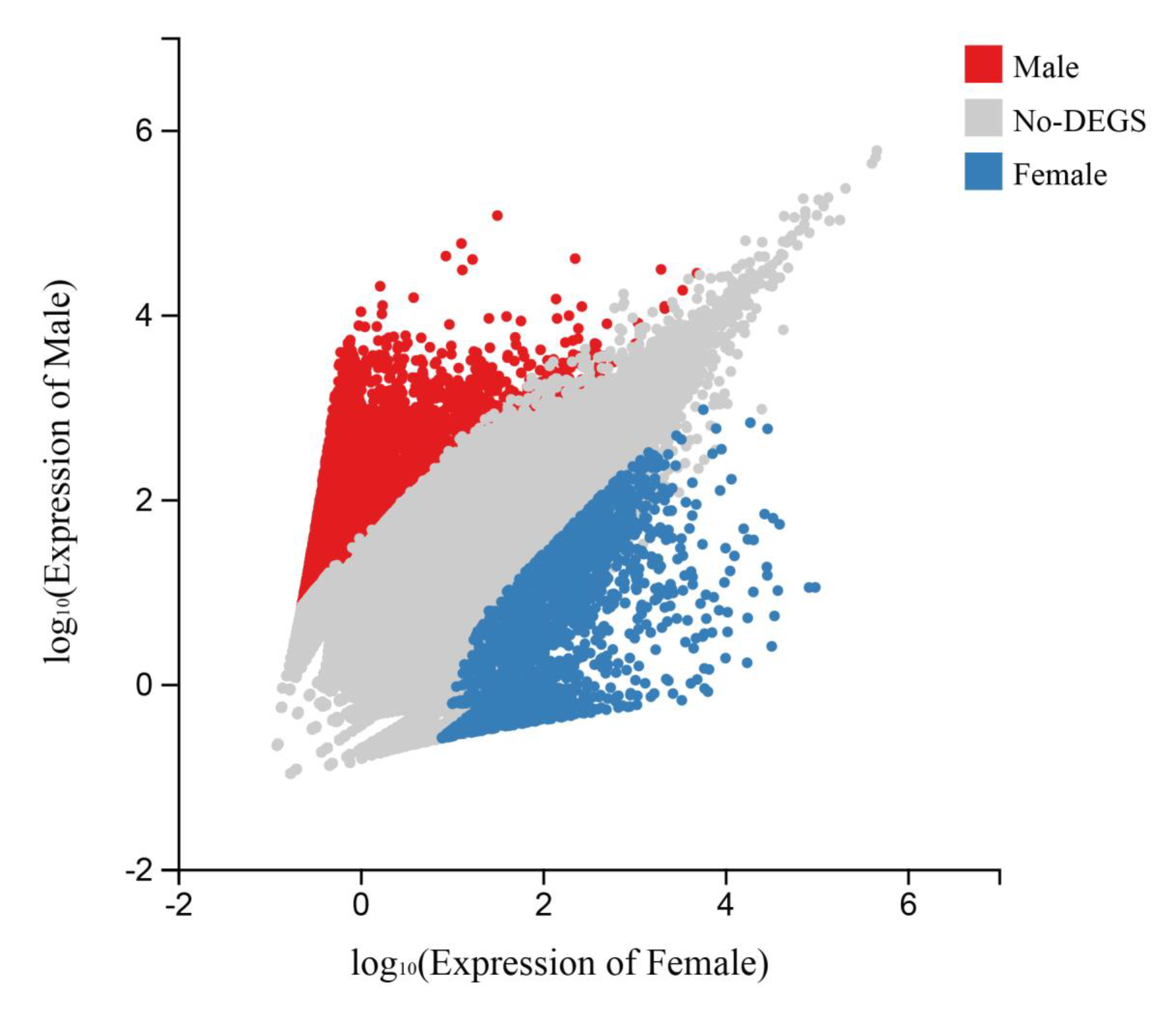
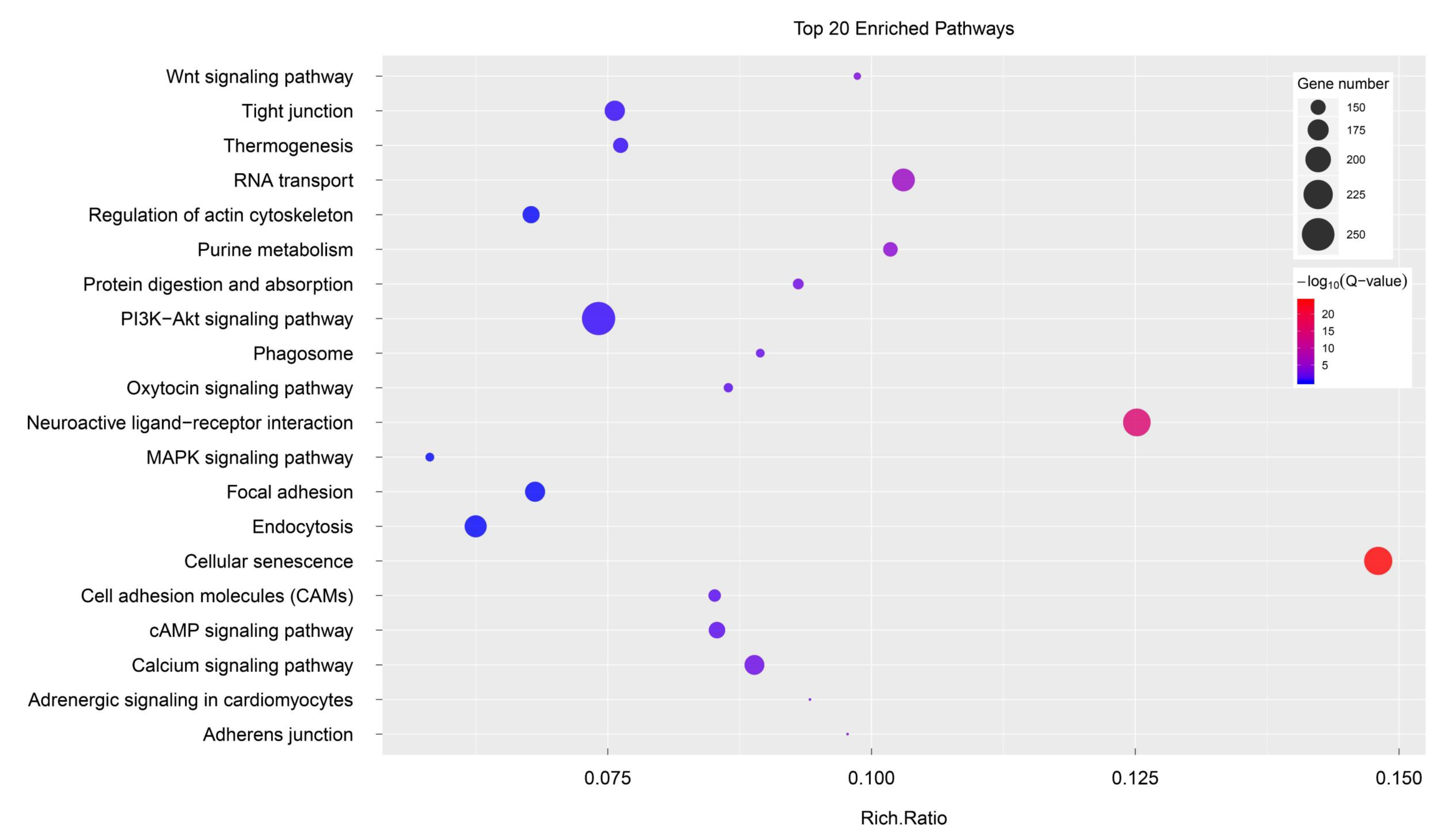
3.3. Potential Candidate Genes and Pathways
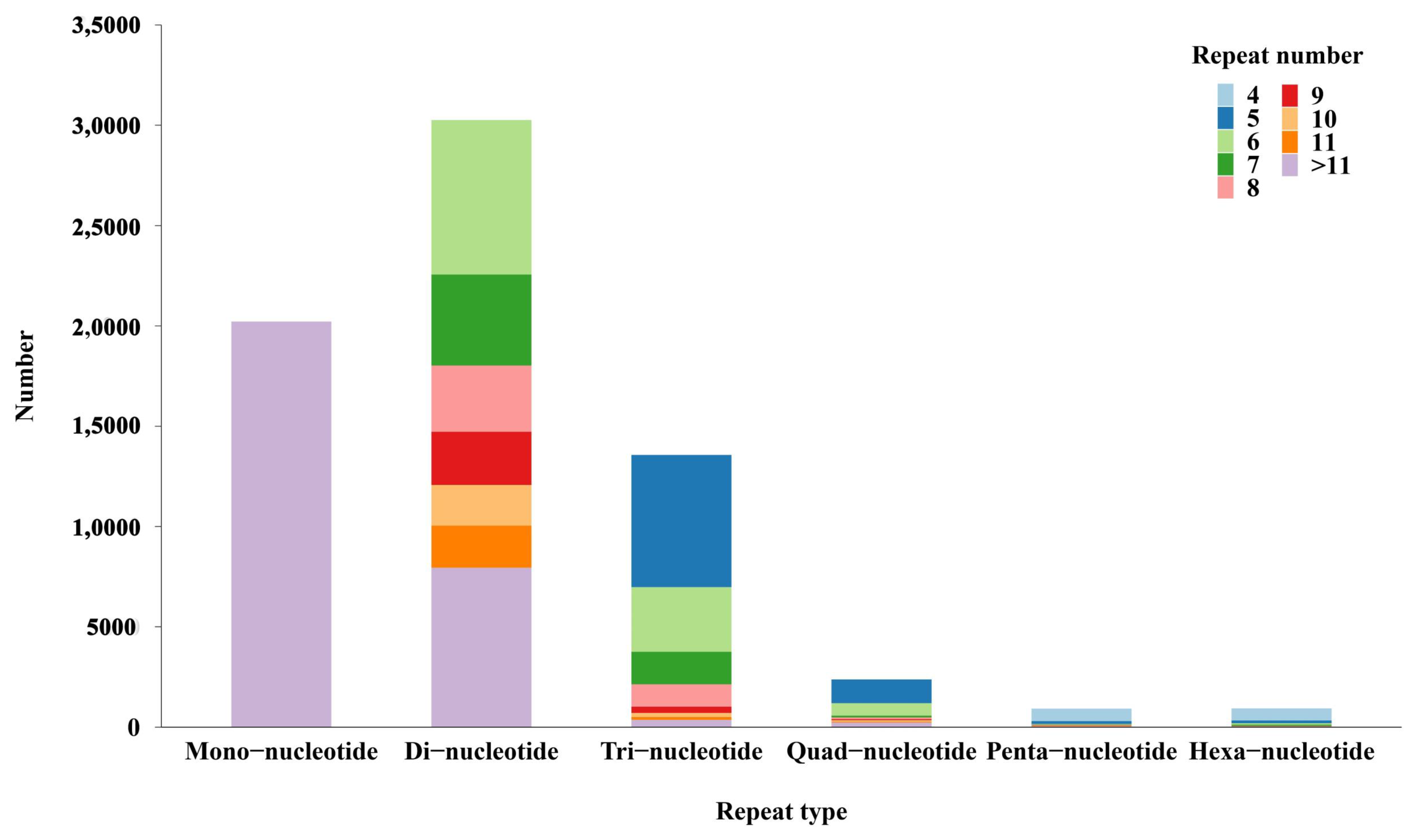
3.4. Discovery of Molecular Markers
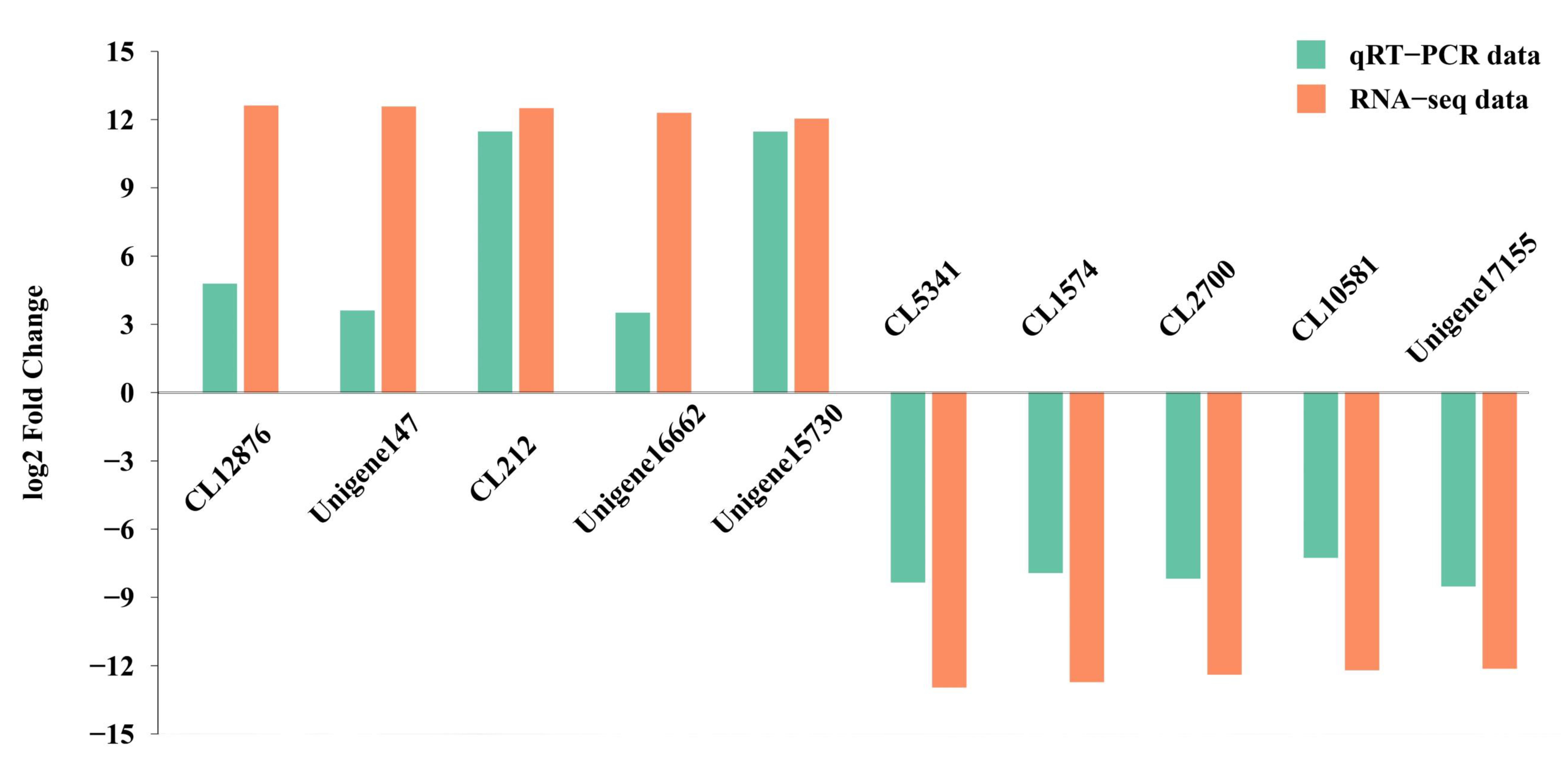
3.5. Transcriptome Data Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Isaac, J.L. Potential causes and life-history consequences of sexual size dimorphism in mammals. Mammal. Rev. 2005, 35, 101–115. [Google Scholar] [CrossRef]
- Loonstra, A.J.; Verhoeven, M.A.; Piersma, T. Sex-specific growth in chicks of the sexually dimorphic Black-tailed Godwit. Ibis 2018, 160, 89–100. [Google Scholar] [CrossRef]
- Pauly, D. Female fish grow bigger-let’s deal with it. Trends Ecol. Evol. 2019, 34, 181–182. [Google Scholar] [CrossRef]
- Toguyeni, A.; Fauconneau, B.; Boujard, T.; Fostier, A.; Kuhn, E.R.; Mol, K.A.; Baroiller, J.F. Feeding behaviour and food utilisation in tilapia, Oreochromis niloticus: Effect of sex ratio and relationship with the endocrine status. Physiol. Behav. 1997, 62, 273–279. [Google Scholar] [CrossRef]
- Simco, B.A.; Goudie, C.A.; Klar, G.T.; Parker, N.C.; Davies, K.B. Influence of sex on growth of channel catfish. Trans. Am. Fish. Soc. 1989, 118, 427–434. [Google Scholar] [CrossRef]
- Haffray, P.; Vauchez, C.; Vandeputte, M.; Linhart, O. Different growth and processing traits in males and females of European catfish, Silurus glanis. Aquat. Living Resour. 1998, 11, 341–345. [Google Scholar] [CrossRef]
- Quinn, T.P.; Foote, C.J. The effects of body size and sexual dimorphism on the reproductive behavior of sockeye salmon, Oncorhynchus nerka. Anim. Behav. 1994, 48, 751–761. [Google Scholar] [CrossRef]
- Bonnet, S.; Haffray, P.; Blanc, J.M.; Vallée, F.; Vauchez, C.; Fauré, A.; Fauconneau, B. Genetic variation in growth parameters until commercial size in diploid and triploid freshwater rainbow trout (Oncorhynchus mykiss) and seawater brown trout (Salmo trutta). Aquaculture 1999, 173, 359–375. [Google Scholar] [CrossRef]
- Imsland, K.; Folkvord, A.; Grung, G.L.; Stefansson, S.O.; Taranger, G.L. Sexual dimorphism in growth and maturation of turbot, Scophthalmus maximus (Rafinesque, 1810). Aquac. Res. 1997, 28, 101–114. [Google Scholar] [CrossRef]
- Saillant, E.; Fostier, A.; Menu, B.; Haffray, P.; Chatain, B. Sexual growth dimorphism in sea bass Dicentrarchus labrax. Aquaculture 2001, 202, 371–387. [Google Scholar] [CrossRef]
- Roncarati, P.; Melotti, O.; Mordenti, L.; Gennari, L. Influence of stocking density of European eel (Anguilla anguilla, L) elvers on sex differentiation and zootechnical performances. J. Appl. Ichtyol. 1997, 13, 131–136. [Google Scholar] [CrossRef]
- Tyus, H.M. Ecology and management of Colorado squawfish. In Battle Against Extinction; Minckley, W.L., Ed.; Native Fish Management in the American West: Tucson, AZ, USA, 1993; pp. 379–402. [Google Scholar]
- Dutney, L.; Elizur, A.; Lee, P. Analysis of sexually dimorphic growth in captive reared cobia (Rachycentron canadum) and the occurrence of intersex individuals. Aquaculture 2017, 468, 348–355. [Google Scholar] [CrossRef]
- Pongthana, N.; Penman, D.J.; Baoprasertkul, P.; Hussain, M.G.; Islam, M.S.; Powell, S.F.; McAndrew, B.J. Monosex female production in the silver barb (Puntius gonionotus Bleeker). Aquaculture 1999, 173, 247–256. [Google Scholar] [CrossRef]
- Rennie, M.D.; Purchase, C.F.; Lester, N.; Collins, N.C.; Shuter, B.J.; Abrams, P.A. Lazy males? Bioenergetic differences in energy acquisition and metabolism help to explain sexual size dimorphism in percids. J. Anim. Ecol. 2008, 77, 916–926. [Google Scholar] [CrossRef]
- Wang, N.; Wang, R.; Wang, R.; Chen, S. Transcriptomics analysis revealing candidate networks and genes for the body size sexual dimorphism of Chinese tongue sole (Cynoglossus semilaevis). Funct. Integr. Genom. 2018, 18, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Lou, F.; Yang, T.; Han, Z.; Gao, T. Transcriptome analysis for identification of candidate genes related to sex determination and growth in Charybdis japonica. Gene 2018, 677, 10–16. [Google Scholar] [CrossRef]
- Ma, D.; Ma, A.; Huang, Z.; Wang, G.; Wang, T.; Xia, D.; Ma, B. Transcriptome analysis for identification of genes related to gonad differentiation, growth, immune response and marker discovery in the turbot (Scophthalmus maximus). PLoS ONE 2016, 11, e0149414. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Zhang, Z.; He, S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet Fish Gymnodiptychus pachycheilus. Genome Biol. Evol. 2015, 7, 251–261. [Google Scholar] [CrossRef]
- Chen, H.; Xiao, G.; Chai, X.; Lin, X.; Fang, J.; Teng, S. Transcriptome analysis of sex-related genes in the blood clam Tegillarca granosa. PLoS ONE 2017, 12, e0184584. [Google Scholar] [CrossRef]
- Koopman, P.; Gubbay, J.; Vivian, N.; Goodfellow, P.; Lovell-Badge, R. Male development of chromosomally female mice transgenic for Sry. Nature 1991, 351, 117–121. [Google Scholar] [CrossRef]
- Hattori, R.S.; Murai, Y.; Oura, M.; Masuda, S.; Majhi, S.K.; Sakamoto, T.; Fernandino, J.I.; Somoza, G.M.; Yokota, M.; Strüssmann, C.A. AY-linked anti-Mullerian hormone duplication takes over a critical role in sex determination. Proc. Natl. Acad. Sci. USA 2012, 109, 2955–2959. [Google Scholar] [CrossRef]
- Yano, A.; Guyomard, R.; Nicol, B.; Jouanno, E.; Quillet, E.; Klopp, C.; Cabau, C.; Bouchez, O.; Fostier, A.; Guiguen, Y. An immune-related gene evolved into the master sex-determining gene in rainbow trout, Oncorhynchus mykiss. Curr. Biol. 2012, 22, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Dan, C.; Lin, Q.; Gong, G.; Yang, T.; Xiong, S.; Xiong, Y.; Huang, P.; Gui, J.-F.; Mei, J. A novel PDZ domain-containing gene is essential for male sex differentiation and maintenance in yellow catfish (Pelteobagrus fulvidraco). Sci. Bull. 2018, 63, 1420–1430. [Google Scholar] [CrossRef]
- Masuda, Y.; Shinohara, N.; Takahashi, Y.; Tabeta, O.; Matsuura, K. Occurrence of natural hybrid between pufferfishes, Takifugu xanthopterus and T. vermicularis, in Ariake Bay, Kyushu, Japan. Nippon. Suisan Gakkaishi 1991, 57, 1247–1255. [Google Scholar] [CrossRef][Green Version]
- Miyaki, K.; Tabeta, O.; Kayano, H. Karyotypes in six species of pufferfishes genus Takifugu (Tetraodontidae, Tetraodontiformes). Fish. Sci. 1995, 61, 594–598. [Google Scholar] [CrossRef]
- Kikuchi, K.; Iwata, N.; Furuta, T.; Kawabata, T.; Yanagawa, T. Growth of tiger puffer Takifugu rubripes in closed recirculating culture system. Fish. Sci. 2006, 72, 1042–1047. [Google Scholar] [CrossRef]
- Wang, Q.L.; Zhang, H.T.; Ren, Y.Q.; Zhou, Q. Comparison of growth parameters of tiger puffer Takifugu rubripes from two culture systems in China. Aquaculture 2016, 453, 49–53. [Google Scholar] [CrossRef]
- Masuda, H.; Amaoka, K.; Muzik, C.K.; Uyeno, T.T.; Yoshimo, T. The Fishes of the Japanese Archipelago; Tokai University Press: Tokyo, Japan, 1984; p. 437. [Google Scholar]
- Yang, Z.; Chen, Y. Length-weight relationship of obscure puffer (Takifugu obscurus) during spawning migration in the Yangtze River, China. J. Freshw. Ecol. 2003, 18, 349–352. [Google Scholar] [CrossRef]
- Ueda, Y.; Sano, J.; Uchida, H.; Amano, C.; Matsumura, Y.; Katayama, T. Growth and age-length key of the tiger puffer Takifugu rubripes in the East China Sea, Sea of Japan, and Seto Inland Sea, Japan. Nippon. Suisan Gakkaishi 2010, 76, 803–811. [Google Scholar]
- Zhou, H.; Zhuang, Z.; Zhang, R. Temperature-control-induced masculinization in tiger puffer Takifugu rubripes. J. Oceanol. Limnol. 2019, 37, 1125–1135. [Google Scholar] [CrossRef]
- Chatchaiphan, S.; Srisapoome, P.; Kim, J.H.; Devlin, R.H.; Na-Nakorn, U. De novo transcriptome characterization and growth-related gene expression profiling of diploid and triploid bighead catfish (Clarias macrocephalus Günther 1864). Mar. Biotechnol. 2017, 19, 36–48. [Google Scholar] [CrossRef]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genom. Hum. G. 2009, 10, 135–151. [Google Scholar] [CrossRef]
- Wolf, J.B. Principles of transcriptome analysis and gene expression quantification: An RNA-seq tutorial. Mol. Ecol. Resour. 2013, 13, 559–572. [Google Scholar] [CrossRef]
- Casas, L.; Saborido-Rey, F.; Ryu, T.; Michell, C.; Ravasi, T.; Irigoien, X. Sex change in clownfish: Molecular insights from transcriptome analysis. Sci. Rep. 2016, 6, 35461. [Google Scholar] [CrossRef]
- Yang, X.; Ikhwanuddin, M.; Li, X.; Lin, F.; Wu, Q.; Zhang, Y.; You, C.; Liu, W.; Cheng, Y.; Shi, X.; et al. Comparative transcriptome analysis provides insights into differentially expressed genes and long non-coding RNAs between ovary and testis of the mud crab (Scylla paramamosain). Mar. Biotechnol. 2018, 20, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Fisheries Resources and Fisheries Science; China Ocean Press: Beijing, China, 2008; pp. 57–58. [Google Scholar]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- De-Santis, C.; Jerry, D.R. Candidate growth genes in finfish-Where should we be looking? Aquaculture 2007, 272, 22–38. [Google Scholar] [CrossRef]
- Jia, Y.; Jing, Q.; Xing, Z.; Gao, X.; Zhai, J.; Guan, C.; Huang, B. Effects of two different culture systems on the growth performance and physiological metabolism of tiger pufferfish (Takifugu rubripes). Aquaculture 2018, 495, 267–272. [Google Scholar] [CrossRef]
- Jia, Y.; Jing, Q.; Zhai, J.; Guan, C.; Huang, B. Alternations in oxidative stress, apoptosis, and innate-immune gene expression at mRNA levels in subadult tiger puffer (Takifugu rubripes) under two different rearing systems. Fish. Shellfish. Immunol. 2019, 92, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Hanhoff, T.; Lucke, C.; Spener, F. Insights into binding of fatty acids by fatty-acid binding proteins. Mol. Cell. Biochem. 2002, 239, 45–54. [Google Scholar] [CrossRef]
- Kanda, T.; Iseki, S.; Hitomi, M.; Kimura, H.; Odani, S.; Kondo, H.; Matsubara, Y.; Muto, T.; Ono, T. Purification and characterization of a fatty-acid-binding protein from the gastric mucosa of rats: Possible identity with heart fatty-acid-binding protein and its parietal cell localization. Eur. J. Biochem. 1989, 185, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Börchers, T.; Spener, F. Fatty acid binding proteins. Curr. Top. Membr. 1994, 40, 261–294. [Google Scholar]
- Pelsers, M.M.; Hermens, W.T.; Glatz, J.F. Fatty acid-binding proteins as plasma markers of tissue injury. Clin. Chim. Acta 2005, 352, 15–35. [Google Scholar] [CrossRef]
- Ockner, R.K.; Manning, J.A.; Poppenhausen, R.B.; Ho, W.K. A binding protein for fatty acids in cytosol of intestinal mucosa, liver, myocardium, and other tissues. Science 1972, 177, 56–58. [Google Scholar] [CrossRef]
- Stewart, J.M.; Driedzic, W.R. Fatty acid binding proteins in teleost fish. Can. J. Zool. 1988, 66, 2671–2675. [Google Scholar] [CrossRef]
- Londraville, R.L.; Sidell, B.L. Cold acclimation increases fatty acidbinding protein concentration in aerobic muscle of striped bass, Morone saxatilis. J. Exp. Zool. 1996, 275, 36–44. [Google Scholar] [CrossRef]
- Ando, S.; Xue, X.H.; Tibbits, G.F.; Haunerland, N.H. Cloning and sequencing of complementary DNA for fatty acid binding protein from rainbow trout heart. Comp. Biochem. Physiol. B 1998, 119, 213–217. [Google Scholar] [CrossRef]
- Jordal, A.E.O.; Hordvik, I.; Pelsers, M.; Bernlohr, D.A.; Torstensen, B.E. FABP3 and FABP10 in Atlantic salmon (Salmo salar L.)-General effects of dietary fatty acid composition and life cycle variations. Comp. Biochem. Physiol. B 2006, 145, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lücke, C.; Gutiérrez-González, L.H.; Hamilton, J.A. Intracellular lipid binding proteins: Evolution, structure, and ligand binding. In Cellular Proteins and Their Fatty Acids in Health and Disease; Duttaroy, A.S., Ed.; Wiley VCH: Weinheim, Germany, 2003; pp. 95–118. [Google Scholar]
- Bass, N.M. The cellular fatty acid binding proteins: Aspects of structure, regulation, and function. Int. Rev. Cytol. 1988, 3, 143–184. [Google Scholar]
- Binas, B.; Danneberg, H.; McWhir, J.; Mullins, L.; Clark, A.J. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999, 13, 805–812. [Google Scholar] [CrossRef]
- Leaver, M.J.; Boukouvala, E.; Antonopoulou, E.; Diez, A.; Favre-Krey, L.; Ezaz, M.T.; Bautista, J.M.; Tocher, D.R.; Krey, G. Three peroxisome proliferators activated receptor isotypes from each of two species of marine fish. Endocrinology 2005, 146, 3150–3162. [Google Scholar] [CrossRef]
- Wolfrum, C.; Borchers, T.; Sacchettini, J.C.; Spener, F. Binding of fatty acids and peroxisome proliferators to orthologous fatty acid binding proteins from human, murine, and bovine liver. Biochemistry 2000, 39, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.B.; Wright, J.M. Comparative genomic organisation and tissue-specific transcription of the duplicated fabp7 and fabp10 genes in teleost fishes. Genome 2013, 56, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Thirumaran, A.; Wright, J.M. Fatty acid-binding protein (fabp) genes of spotted green pufferfish (Tetraodon nigroviridis): Comparative genomics and spatial transcriptional regulation. Genome 2014, 57, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of FABPs-mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592. [Google Scholar] [CrossRef]
- Ogino, T.; Moralejo, D.H.; Kose, H.; Yamada, T.; Matsumoto, K. Serum leptin concentration is linked to chromosomes 2 and 6 in the OLETF rat, an animal model of type 2 diabetes with mild obesity. Mamm. Genome 2003, 14, 239–244. [Google Scholar] [CrossRef]
- Gerbens, F.; Verburg, F.J.; van Moerkerk, H.T.B.; Engel, B.; Buist, W.; Veerkamp, J.H.; Te Pas, M.F.W. Associations of heart and adipocyte fatty acid-binding protein gene expression with intramuscular fat content in pigs. J. Anim. Sci. 2001, 79, 347–354. [Google Scholar] [CrossRef]
- Mercadé, A.; Pérez-Enciso, M.; Varona, L.; Alves, E.; Noguera, J.L.; Sánchez, A.; Folch, J.M. Adipocyte fatty-acid binding protein is closely associated to the porcine FAT1 locus on chromosome 4. J. Anim. Sci. 2006, 84, 2907–2913. [Google Scholar] [CrossRef]
- Power, D.M. Developmental ontogeny of prolactin and its receptor in fish. Gen. Comp. Endocr. 2005, 142, 25–33. [Google Scholar] [CrossRef]
- Bern, H.A. Functional evolution of prolactin and growth hormone in lower vertebrates. Am. Zool. 1983, 23, 663–671. [Google Scholar] [CrossRef]
- WClarke, C.; Bern, H.A. Comparative endocrinology of prolactin. In Hormonal Proteins and Peptides; Li, C.H., Ed.; Academic Press: New York, NY, USA, 1980; pp. 105–197. [Google Scholar]
- Yang, B.Y.; Greene, M.; Chen, T.T. Early embryonic expression of the growth hormone family protein genes in the developing rainbow trout, Oncorhynchus mykiss. Mol. Reprod. Dev. 1999, 53, 127–134. [Google Scholar] [CrossRef]
- Santos, C.R.A.; Brinca, L.; Ingleton, P.M.; Power, D.M. Cloning, expression, and tissue localisation of prolactin in adult sea bream (Sparus aurata). Gen. Comp. Endocr. 1999, 114, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Kaneko, T.; Aida, K. Prolactin and prolactin receptor expressions in a marine teleost, pufferfish Takifugu rubripes. Gen. Comp. Endocr. 2006, 146, 318–328. [Google Scholar] [CrossRef]
- Shepherd, B.S.; Sakamoto, T.; Nishioka, R.S.; Richman, N.H.; Mori, I.; Madsen, S.S.; Chen, T.T.; Hirano, T.; Bern, H.A.; Grau, E.G. Somatotropic actions of the homologous growth hormone and prolactins in the euryhaline teleost, the tilapia, Oreochromis mossambicus. Proc. Natl. Acad. Sci. USA 1997, 94, 2068–2072. [Google Scholar] [CrossRef]
- Ribas, L.; Robledo, D.; Gómez-Tato, A.; Viñas, A.; Martínez, P.; Piferrer, F. Comprehensive transcriptomic analysis of the process of gonadal sex differentiation in the turbot (Scophthalmus maximus). Mol. Cell Endocrinol. 2016, 422, 132–149. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Nagahama, Y.; Nakamura, M. Diversity and plasticity of sex determination and differentiation in fishes. Sex. Dev. 2013, 7, 115–125. [Google Scholar] [CrossRef]
- Du, X.; Wang, B.; Liu, X.; Liu, X.; He, Y.; Zhang, Q.; Wang, X. Comparative transcriptome analysis of ovary and testis reveals potential sex-related genes and pathways in spotted knifejaw Oplegnathus punctatus. Gene 2017, 637, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, Y.; Wang, W.; Wang, Q.; Zhang, N.; Lin, F.; Wang, N.; Shao, C.; Dong, Z.; Li, Y.; et al. Genome editing reveals dmrt1 as an essential male sex-determining gene in Chinese tongue sole (Cynoglossus semilaevis). Sci. Rep. 2017, 7, 42213. [Google Scholar] [CrossRef]
- Webster, K.A.; Schach, U.; Ordaz, A.; Steinfeld, J.S.; Draper, B.W.; Siegfried, K.R. Dmrt1 is necessary for male sexual development in zebrafish. Dev. Biol. 2017, 422, 33–46. [Google Scholar] [CrossRef]
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 2000, 227, 239–255. [Google Scholar] [CrossRef]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Takehana, Y.; Matsuda, M.; Myosho, T.; Suster, M.L.; Kawakami, K.; Shin-I, T.; Kohara, Y.; Kuroki, Y.; Toyoda, A.; Fujiyama, A.; et al. Co-option of Sox3 as the male-determining factor on the Y chromosome in the fish Oryzias dancena. Nat. Commun. 2014, 5, 4157. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhou, L.; Wang, Y.; Xia, W.; Gui, J. Differential expression and dynamic changes of SOX3 during gametogenesis and sex reversal in protogynous hermaphroditic fish. J. Exp. Zool. A Ecol. Genet. Physiol. 2007, 307, 207–219. [Google Scholar] [CrossRef]
- Li, S.; Lin, G.; Fang, W.; Huang, P.; Gao, D.; Huang, J.; Xie, J.; Lu, J. Gonadal transcriptome analysis of sex-related genes in the protandrous yellowfin seabream (Acanthopagrus latus). Front. Genet. 2020, 11, 709. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.N.; Brummett, A.R. Egg envelopes in vertebrates. Dev. Biol. 1985, 1, 235–288. [Google Scholar]
- Wassarman, P.M. Zona pellucida glycoproteins. Annu. Rev. Biochem. 1988, 57, 415–442. [Google Scholar] [CrossRef]
- Litscher, E.S.; Wassarman, P.M. The fish Egg’s zona Pellucida. Curr. Top. Dev. Biol. 2018, 130, 275–305. [Google Scholar]
- Litscher, E.S.; Wassarman, P.M. Egg extracellular coat proteins: From fish to mammals. Histol. Histopathol. 2007, 22, 337–347. [Google Scholar]
- Liu, X.; Wang, H.; Gong, Z. Tandem-repeated zebrafish zp3 genes possess oocyte-specific promoters and are insensitive to estrogen induction1. Biol. Reprod. 2006, 74, 1016–1025. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads (M) | Clean Reads (M) | Clean Bases (Gb) | Clean Reads Q30 (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|
| Female-1 | 52.59 | 48.33 | 7.25 | 87.49 | 91.90 |
| Female-2 | 44.04 | 40.55 | 6.08 | 86.17 | 92.06 |
| Female-3 | 47.33 | 44.00 | 6.60 | 86.04 | 92.98 |
| Male-1 | 47.33 | 43.93 | 6.59 | 86.37 | 92.81 |
| Male-2 | 47.33 | 43.63 | 6.54 | 86.48 | 92.19 |
| Male-3 | 47.33 | 43.58 | 6.54 | 86.18 | 92.09 |
| Sample | Total Number | Total Length | Mean Length | N50 | GC (%) |
|---|---|---|---|---|---|
| Female-1 | 81,249 | 145,145,039 | 1786 | 3128 | 48.91 |
| Female-2 | 72,625 | 118,111,439 | 1626 | 2823 | 49.00 |
| Female-3 | 72,194 | 120,065,634 | 1663 | 2836 | 49.06 |
| Male-1 | 93,484 | 144,668,988 | 1547 | 2725 | 48.62 |
| Male-2 | 95,350 | 142,802,517 | 1497 | 2627 | 48.55 |
| Male-3 | 84,292 | 131,779,534 | 1563 | 2735 | 48.81 |
| All-Unigene | 149,814 | 309,566,984 | 2066 | 3538 | 48.64 |
| Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| Total | 122,719 | 81.91% |
| NT | 70,243 | 46.89% |
| NR | 63,250 | 42.22% |
| SwissProt | 67,197 | 44.85% |
| KEGG | 100,035 | 66.77% |
| KOG | 90,961 | 60.72% |
| GO | 63,518 | 42.40% |
| Gene | Unigene | Annotation | Log2(♂/♀) | Q-Value |
|---|---|---|---|---|
| Dmtr1 | Unigene1322 | doublesex and mab-3-related transcription factor 1 | 6.66 | 2.34 × 10−07 |
| Sox3 | Unigene1791 | SRY-box containing transcription factor 3 | 5.72 | 8.44 × 10−04 |
| Dnali1 | Unigene12399 | axonemal dynein light intermediate polypeptide 1 | 7.63 | 3.40 × 10−07 |
| Ropn1l | CL4645.Contig3 | ropporin-1-like protein | 9.37 | 1.32 × 10−18 |
| Cyp19a | CL5324.Contig8 | cytochrome P450 aromatase | −7.54 | 2.15 × 10−07 |
| Zp1 | CL6076.Contig3 | zona pellucida sperm-binding protein 1 | −12.66 | 1.43 × 10−110 |
| Zp2 | CL8003.Contig1 | zona pellucida sperm-binding protein 2 | −7.70 | 1.84 × 10−20 |
| Zp3 | CL7875.Contig2 | zona pellucida sperm-binding protein 3 | −7.93 | 4.04 × 10−16 |
| Start-7 | CL3580.Contig10 | stAR-related lipid transfer protein 7 | 7.57 | 2.66 × 10−06 |
| Gdf3 | CL9083.Contig1 | growth differentiation factor 3 | −8.90 | 3.99 × 10−14 |
| Gdf9 | CL6347.Contig1 | growth differentiation factor 9 | −9.23 | 1.61 × 10−12 |
| Gtf3a | Unigene10317 | Transcription factor IIIA-like | −2.42 | 2.11 × 10−08 |
| 42sp43 | Unigene8014 | P43 5S RNA-binding protein-like | −7.38 | 6.57 × 10−24 |
| Mnd1 | Unigene817 | meiotic nuclear division protein 1 homolog | 3.25 | 8.39 × 10−03 |
| Star | Unigene31813 | steroidogenic acute regulatory protein | −2.49 | 3.42 × 10−04 |
| Wee1 | CL12054.Contig1 | wee1-like protein kinase | 2.05 | 6.42 × 10−03 |
| Spaca6 | CL4045.Contig1 | sperm acrosome membrane-associated protein 6 | 5.97 | 4.05 × 10−04 |
| Spaca4 | CL12530.Contig2 | sperm acrosome membrane-associated protein 4 | 3.53 | 2.18 × 10−15 |
| Spata5 | CL8799.Contig2 | spermatogenesis-associated protein5 | 6.47 | 7.85 × 10−05 |
| Spata7 | CL4586.Contig1 | spermatogenesis-associated protein 7 | 2.93 | 1.40 × 10−03 |
| Spata17 | CL4320.Contig15 | spermatogenesis-associated protein 17 | 8.17 | 2.09 × 10−08 |
| Spata32 | Unigene13081 | spermatogenesis-associated protein 32 | 4.78 | 1.72 × 10−06 |
| Gene | Unigene | Annotation | Log2(♂/♀) | Q-Value |
|---|---|---|---|---|
| Igfbp1 | CL6482.Contig4 | insulin-like growth factor-binding protein 1 | −8.52 | 2.51 × 10−09 |
| Igfbp3 | Unigene7287 | Insulin-like growth factor-binding protein 3 | −3.36 | 9.25 × 10−06 |
| Igf1r | Unigene24558 | Insulin-like growth factor 1 receptor | −1.06 | 1.75 × 10−02 |
| Ghr | CL9297.Contig1 | Growth hormone receptor | −4.79 | 3.10 × 10−03 |
| Sstr2 | Unigene6248 | Somatostatin receptor 2 | 7.45 | 5.93 × 10−07 |
| Fabp1 | CL7463.Contig1 | fatty acid-binding protein 1 | 3.85 | 8.11 × 10−12 |
| Fabp4 | CL10654.Contig1 | fatty acid-binding protein 4 | −8.28 | 6.17 × 10−56 |
| Fabp6 | CL6561.Contig1 | fatty acid-binding protein 6 | 6.70 | 2.41 × 10−04 |
| Fabp7 | Unigene40265 | fatty acid-binding protein 7 | 5.45 | 2.86 × 10−05 |
| Grb1 | CL5906.Contig1 | Growth factor receptor-bound proteins 1 | 2.16 | 6.38 × 10−03 |
| Grb7 | Unigene6338 | Growth factor receptor-bound proteins 7 | −2.98 | 8.15 × 10−03 |
| Grb10 | CL11116.Contig1 | Growth factor receptor-bound proteins 10 | 3.07 | 1.43 × 10−04 |
| Htr1 | Unigene31616 | 5-hydroxytryptamine receptor 1D-like | 4.59 | 1.31 × 10−04 |
| Htr2 | Unigene28993 | 5-hydroxytryptamine receptor 2C-like | 5.27 | 3.86 × 10−03 |
| Htr3 | CL1712.Contig2 | 5-hydroxytryptamine receptor 3 | 5.61 | 1.44 × 10−03 |
| Htr6 | Unigene18275_ | 5-hydroxytryptamine receptor 6 | 4.33 | 7.54 × 10−03 |
| Prlr | CL1256.Contig1 | Prolactin receptor | −8.19 | 3.29 × 10−11 |
| Prl | CL12326.Contig4 | Prolactin | −5.65 | 4.17 × 10−06 |
| Myhc | Unigene12897 | myosin heavy chain | 6.29 | 5.15 × 10−05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, B.; Liu, Y.; Yang, C.; Li, Y.; Wang, L.; Sun, D. Comparative Transcriptome Analysis of Female and Male Fine-Patterned Puffer: Identification of Candidate Genes Associated with Growth and Sex Differentiation. Fishes 2021, 6, 79. https://doi.org/10.3390/fishes6040079
Shan B, Liu Y, Yang C, Li Y, Wang L, Sun D. Comparative Transcriptome Analysis of Female and Male Fine-Patterned Puffer: Identification of Candidate Genes Associated with Growth and Sex Differentiation. Fishes. 2021; 6(4):79. https://doi.org/10.3390/fishes6040079
Chicago/Turabian StyleShan, Binbin, Yan Liu, Changping Yang, Yuan Li, Liangming Wang, and Dianrong Sun. 2021. "Comparative Transcriptome Analysis of Female and Male Fine-Patterned Puffer: Identification of Candidate Genes Associated with Growth and Sex Differentiation" Fishes 6, no. 4: 79. https://doi.org/10.3390/fishes6040079
APA StyleShan, B., Liu, Y., Yang, C., Li, Y., Wang, L., & Sun, D. (2021). Comparative Transcriptome Analysis of Female and Male Fine-Patterned Puffer: Identification of Candidate Genes Associated with Growth and Sex Differentiation. Fishes, 6(4), 79. https://doi.org/10.3390/fishes6040079