1. Introduction
Genetic diversity is an essential component of biodiversity [
1]. Research into the genetic diversity of species not only aids in our understanding of germplasm resources but also provides scientific solutions for species conservation and utilization [
2]. Furthermore, such research has significant implications for selective breeding and genetic improvement [
3]. Key issues for developing management strategies include genetic diversity, population genetic structure, and gene flow [
4,
5]. The genetic structure of populations is shaped by the interplay of ecological and genetic factors. Population genetic structure refers to the distribution of genetic variation within and among populations, shaped by evolutionary processes such as genetic drift, migration, mutation, and selection. It is distinct from genetic diversity, which quantifies the variability of alleles within a population, and gene flow, which describes the movement of genetic material between populations [
6]. Investigating population genetic structure and gene flow is essential for understanding genetic characteristics and population dynamics, providing critical insights for conservation planning for rare and endangered species with restricted geographical distributions [
7].
Mitochondrial DNA (mtDNA) serves as an excellent candidate marker, proving suitable for studies of population genetics and molecular systematics [
8]. The mitochondrial D-loop region has been shown to have more variable sequences than other regions of mtDNA [
9]. Cox1, also known as cytochrome oxidase subunit I, has a moderate rate of evolution [
10]. The mitochondrial genome is maternally inherited [
11]. Consequently, genetic diversity and population structure studies in fish species have integrated the results of analyses based on the mtDNA D-loop and cox1 genes [
12,
13,
14,
15]. The sinipercine subfamily, including species such as
Siniperca chuatsi,
Siniperca scherzeri, and
Coreoperca whiteheadi, is important both ecologically and economically in East Asia. Previous genetic studies have focused on mitochondrial DNA to understand their diversity and phylogenetic relationships. The complete mitochondrial genome of
Siniperca obscura (16,492 bp) was sequenced, providing insights into the species’ phylogeny [
16]. Combined analyses of mitochondrial and microsatellite markers in
S. scherzeri revealed distinct evolutionary lineages with significant regional differentiation, providing valuable insights for conservation and breeding programs [
17]. Furthermore, the mitochondrial genome of a hybrid between
S. chuatsi and
S. kneri was sequenced, offering valuable insights into the genetic mechanisms underlying hybridization in Sinipercinae [
18].
The aquaculture industry has developed rapidly in recent years [
19]. Unfortunately, the accelerated intensification of aquaculture production has simultaneously compromised both natural resource sustainability and genetic diversity conservation. [
20]. Mandarin fish (
Siniperca chuatsi), a member of the Sinipercinae, is native to China and is one of the most important freshwater economic fishes in the country [
21]. It is also distributed in Vietnam, Japan, the Korean peninsula, and the Russian Far East [
22,
23]. Due to its rapid growth and high meat quality,
S. chuatsi has been cultured in many provinces in China [
24]. However, artificiality bred populations typically exhibit reduced genetic diversity due to selective breeding for commercially desirable traits, including accelerated growth rates, enhanced meat production, and improved resistance to both pathogens and environmental stressors. [
25,
26,
27]. The golden mandarin fish (
S. scherzeri) and
C. whiteheadi, both members of the subfamily Sinipercinae, are widely distributed freshwater species native to China, and are also found in Vietnam and the Korean peninsula [
22,
23].
S. scherzeri is often crossed with
S. chuatsi because of its various important economic traits, such as acceptance of minced fish and resistance to environmental stress [
28]. However, the natural resources of the other two sinipercine species have declined dramatically in recent years due to anthropogenic interference and overexploitation [
17]. Thus, the analysis of genetic diversity among the three sinipercine species, particularly for
S. chuatsi and
S. scherzeri, is crucial not only for understanding their evolutionary history but also for informing long-term resource management and sustainable use strategies.
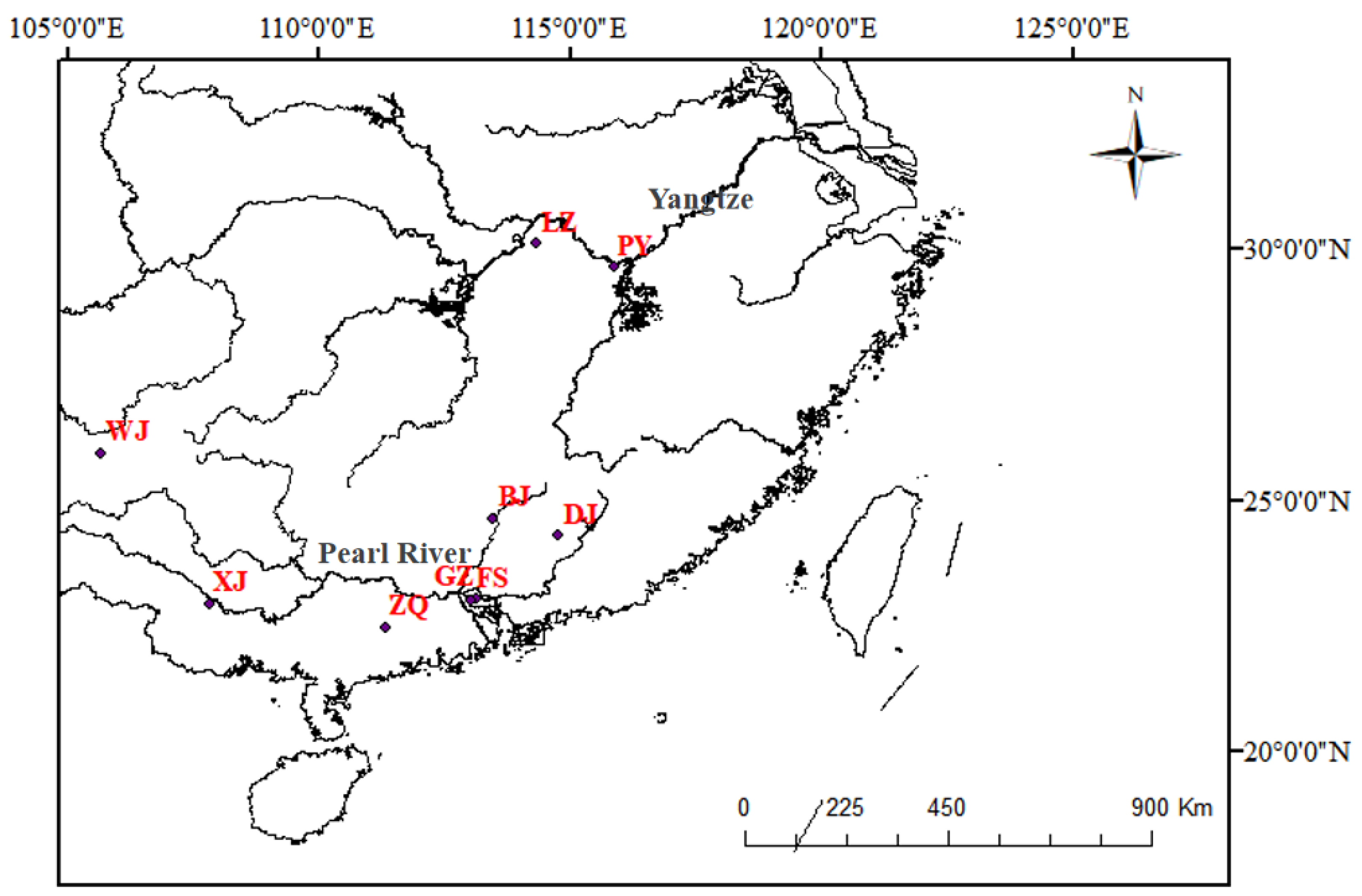
In this study, we aimed to analyze the population genetic structure of S. chuatsi and S. scherzeri in China. To achieve this, we examined the population subdivision and genetic variability of S. chuatsi across five populations and S. scherzeri across three populations using the D-loop region and the cox1 gene. Additionally, we characterized the phylogenetic relationships within S. chuatsi populations and assessed the relationships between S. chuatsi, the wild species S. scherzeri, and C. whiteheadi. The results of this study provide valuable insights for the conservation and sustainable management of S. chuatsi resources and supporting artificial hybridization efforts among these economically important fish species.
4. Discussion
Despite its nutritional and economic importance,
S. chuatsi genetic resources and artificial hybridization with other sinipercine fishes have only rarely been reported [
41,
42]. In this study, we aimed to measure the genetic diversity and population structure of
S. chuatsi,
S. scherzeri, and
C. whiteheadi to aid senetic resource management and artificial hybridization.
Haplotype diversity (
Hd) and nucleotide diversity (
π) are two important indicators of genetic diversity among species or populations [
43]. We analyzed data from the D-loop region and cox1 sequences in 207 sinipercine fishes (107
S. chuatsi, 93
S. scherzeri, and 7
C. whiteheadi). In most of the sampled populations, the D-loop region exhibited more haplotypes than the cox1 sequences, with the exception of the LZ population. The higher genetic diversity observed in the D-loop region compared to cox1 is consistent with the results of previous studies that documented greater variation in the D-loop region [
14,
44]. Many researchers have used concatenated sequences in analyses to improve the reliability of the results [
13,
36]. We thus used concatenated sequences of the D-loop region and cox1 to further understand genetic diversity and population structure. The five
S. chuatsi populations, namely GZ, FS, ZQ, LZ, and PY, all had a high diversity of haplotypes (
Hd > 0.5), indicating substantial genetic variation. However, the FS, ZQ, and LZ populations exhibited lower nucleotide diversity (
π < 0.005) compared to the GZ and PY populations, reflecting lesser nucleotide variation. Similar results were obtained for the BJ and DJ populations of
S. scherzeri and
C. whiteheadi. This observation is consistent with the results of previous studies of sinipercine fishes [
17,
45]. Genetic diversity is important for selective breeding for market traits and is responsive to the influence of genetic bottlenecks, geographical isolation, and artificial selection. For domesticated animals, we need to be cautious about captive breeding because if we mate offspring indiscriminately, genetic diversity will become limited, and after many generations, genetic diversity can be depleted, resulting in inbreeding. FS cultured populations have lower genetic diversity than wild populations, which might stem from the tendency to have a limited number of broodstock in captive breeding. There is thus a need to improve the genetic management of these species. Compared to the BJ and DJ populations, the WJ population showed high haplotype diversity and high nucleotide diversity. The WJ population’s higher genetic diversity likely reflects lower anthropogenic pressure and greater habitat connectivity compared to those in other watersheds.
Genetic distance analysis indicated that the LZ population was genetically distinct from the other S. chuatsi populations. This could be attributed to the small sample size from LZ, as obtaining samples from this area proved difficult, highlighting the scarcity of wild S. chuatsi in this region. As a result, the sample size for certain populations, such as the S. chuatsi wild group from LZ, was relatively small. Future studies should aim to include a larger and more balanced sample size, especially from regions with limited access.
In addition, the genetic distance was far lower between BJ and DJ than between WJ and S. scherzeri, which indicated that the BJ and DJ populations showed slight genetic differentiation. Another limitation of our study is that the geographic coverage is somewhat restricted, as sampling was concentrated in specific regions. Furthermore, we examined mtDNA variation, but did not examine nuclear markers. A broader sampling approach that includes populations from a wider range of environmental conditions and geographical locations would likely provide a more comprehensive understanding of the genetic diversity of these species. Temporal coverage is also a factor; future research should consider sampling over multiple seasons to account for temporal variations in genetic diversity of samples.
FST can be used to estimate gene exchange and genetic differentiation between populations [
46].
FST, an important population differentiation index, revealed significant genetic differentiation between most
S. chuatsi and
S. scherzeri populations. According to Wright [
46], genetic differentiation between populations is high when 0.15 <
FST < 0.25. Similarly, significant genetic differentiation has been reported in many fish species, reinforcing the point that watershed isolation drives divergence more strongly than geographic distance alone [
47,
48,
49,
50]. Geographical isolation, dwindling wild resources, and the effects of captive breeding could result in a higher
FST of FS and two wild populations, PY and LZ. The
FST of FS and GZ was higher, while the
FST of ZQ was lower than the other captively bred stocks. This may be related to the ZQ stock’s diverse sources and lack of population management, which could cause issues in genetic improvement. The observed genetic diversity in captive populations may be influenced by multiple factors, including artificial selection practices and potentially random genetic drift. While our current dataset cannot definitively quantify their relative contributions, we acknowledge that genetic drift may play a non-negligible role, particularly in smaller breeding populations. This important consideration warrants further investigation in future studies.
The estimated gene flow was high among most
S. chuatsi populations (
Nm > 1) (
Table 3); three wild
S. scherzeri populations also showed high gene flow (
Nm > 1) (
Table 4).
Nm is inversely proportional to
FST and proportional to genetic diversity. The GZ and ZQ cultured populations had small
FST (0.111) and high
Nm (4.01). These two populations are close in proximity and have access to a greater quantity of wild resources. The farms’ wild supplementary parents might be from the same resource population, which means that these distinct groups might share genetic heritage. Gene flow between the
S. scherzeri neighboring wild populations BJ and DJ is not limited, according to an analysis of
FST and estimated gene flow.
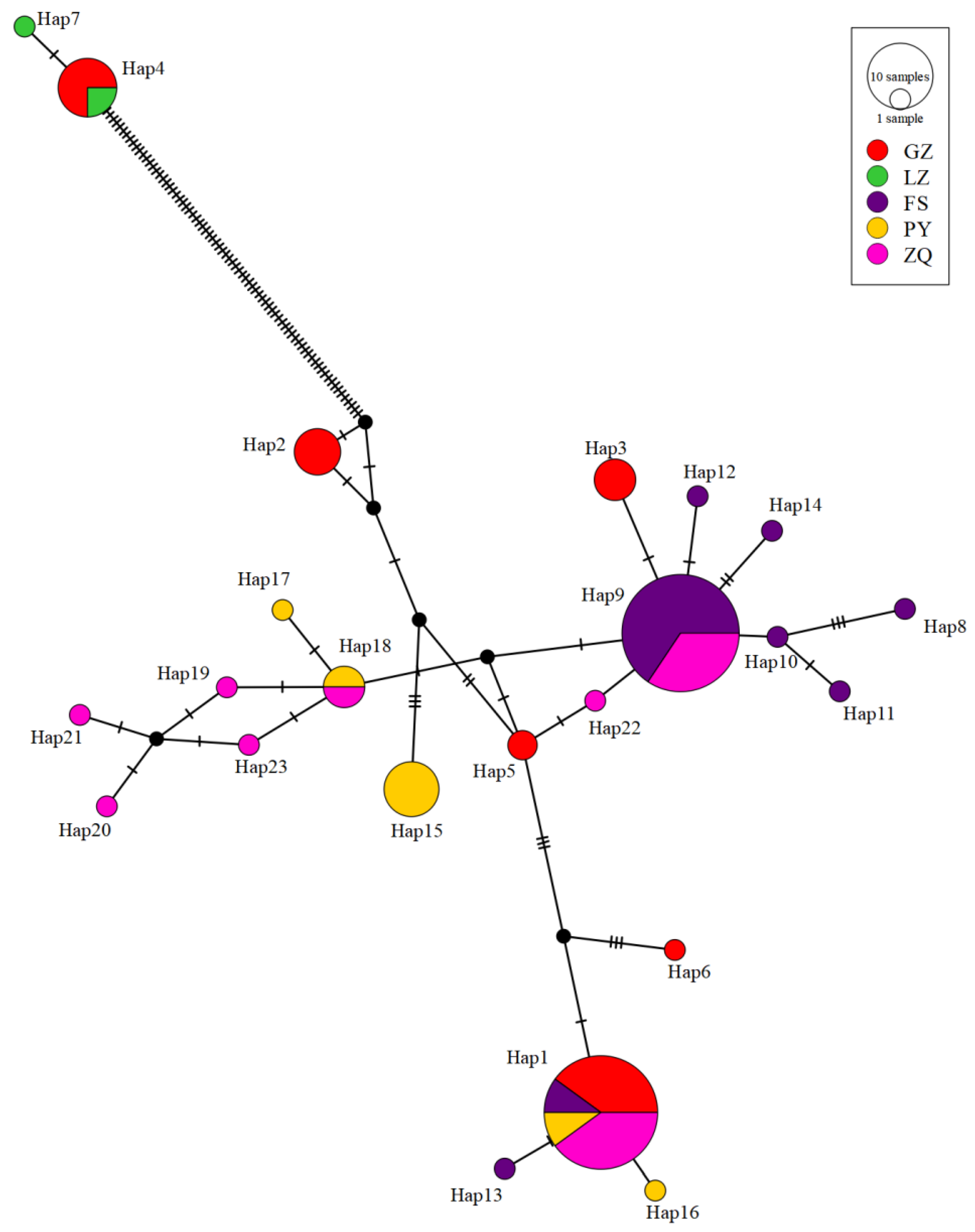
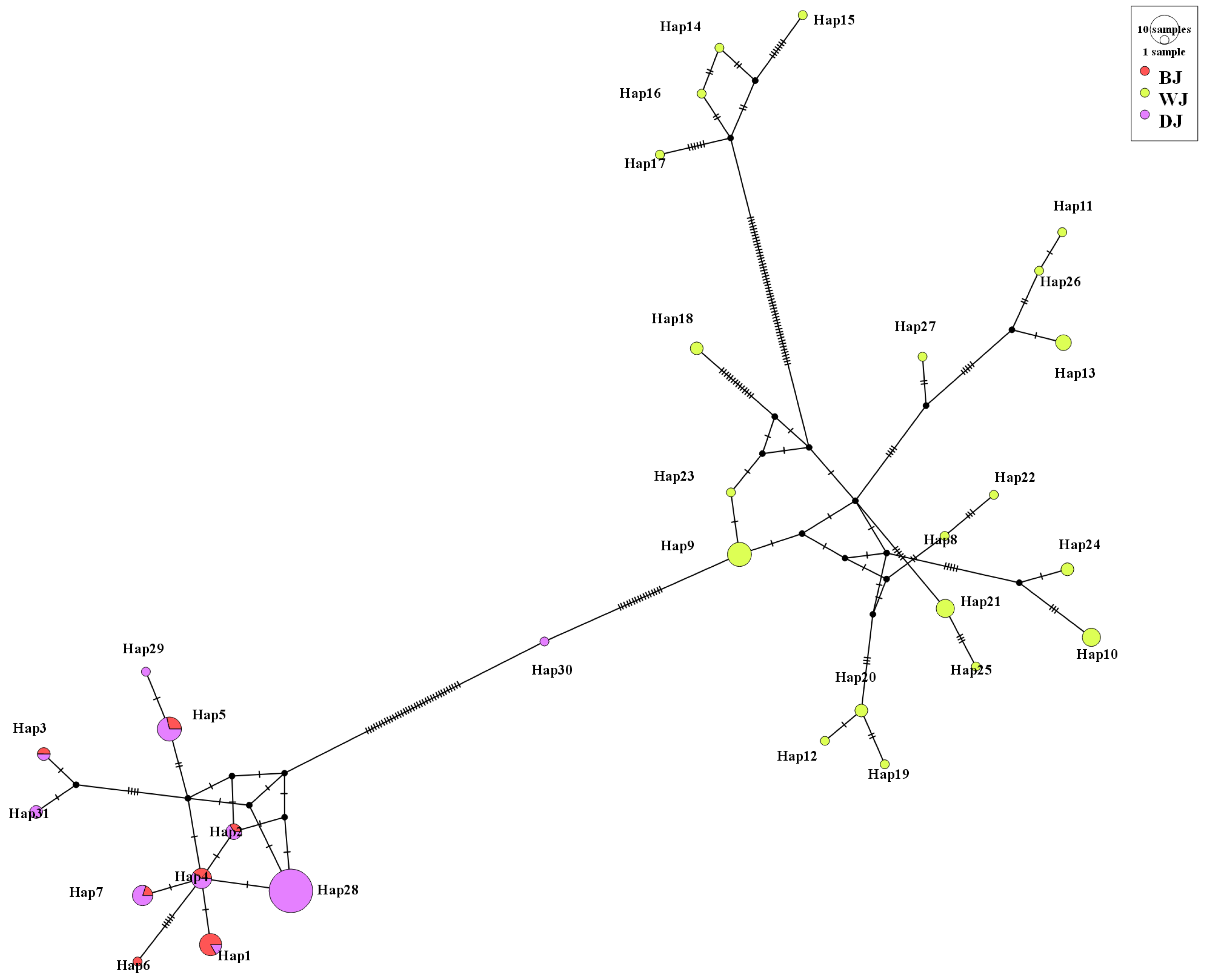
Based on the phylogenetic analysis and haplotype network in S. chuatsi, four haplotypes were shared between cultured and wild populations, especially in ZQ. The shared haplotypes between cultured and wild populations suggest either recent wild broodstock use or limited genetic divergence under culture. This finding is consistent with the results of AMOVA analysis and FST. Possible anthropogenic explanations for shared haplotypes include the occasional introduction of wild broodstock into farms, commercial trade facilitating genetic mixing between regions, and habitat degradation in source watersheds promoting genetic drift that reduces haplotype diversity in wild populations subsequently used for aquaculture. Phylogenetic and haplotype network analyses of S. scherzeri revealed two distinct clades among the three populations. BJ and DJ shared six common haplotypes, while WJ had none, likely because WJ occupies a different watershed.
These findings highlight the need for improved management of wild Siniperca stocks, including regulations on translocations between watersheds to preserve natural genetic structure. For aquaculture, our results suggest that maintaining genetic diversity in farmed populations requires controlled breeding strategies and periodic genetic monitoring. This study provides a foundation for developing science-based conservation and farming practices for these economically important species.




{kind=link}
{kind=link}
{kind=link}
{kind=link}