
Genetic Diversity and Population Structure of Commercial Eel Conger myriaster (Anguilliformes: Congridae) Along the Coasts of China Based on Complete Mitochondrial Cyt b Sequences


Abstract
1. Introduction
2. Materials and Methods
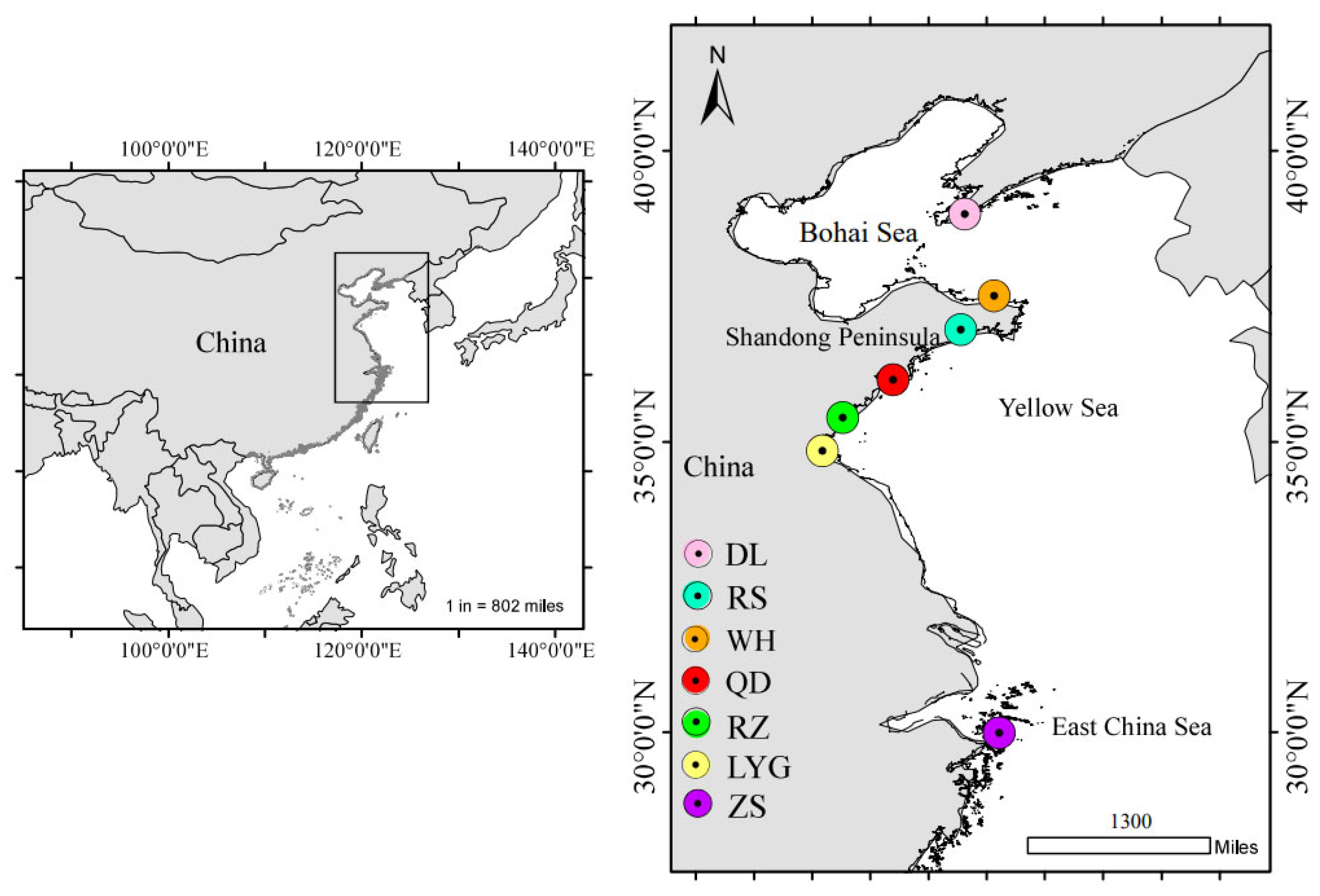
2.1. Sampling, DNA Extraction and PCR Amplification
2.2. Data Analysis
3. Results
3.1. Sequence Information
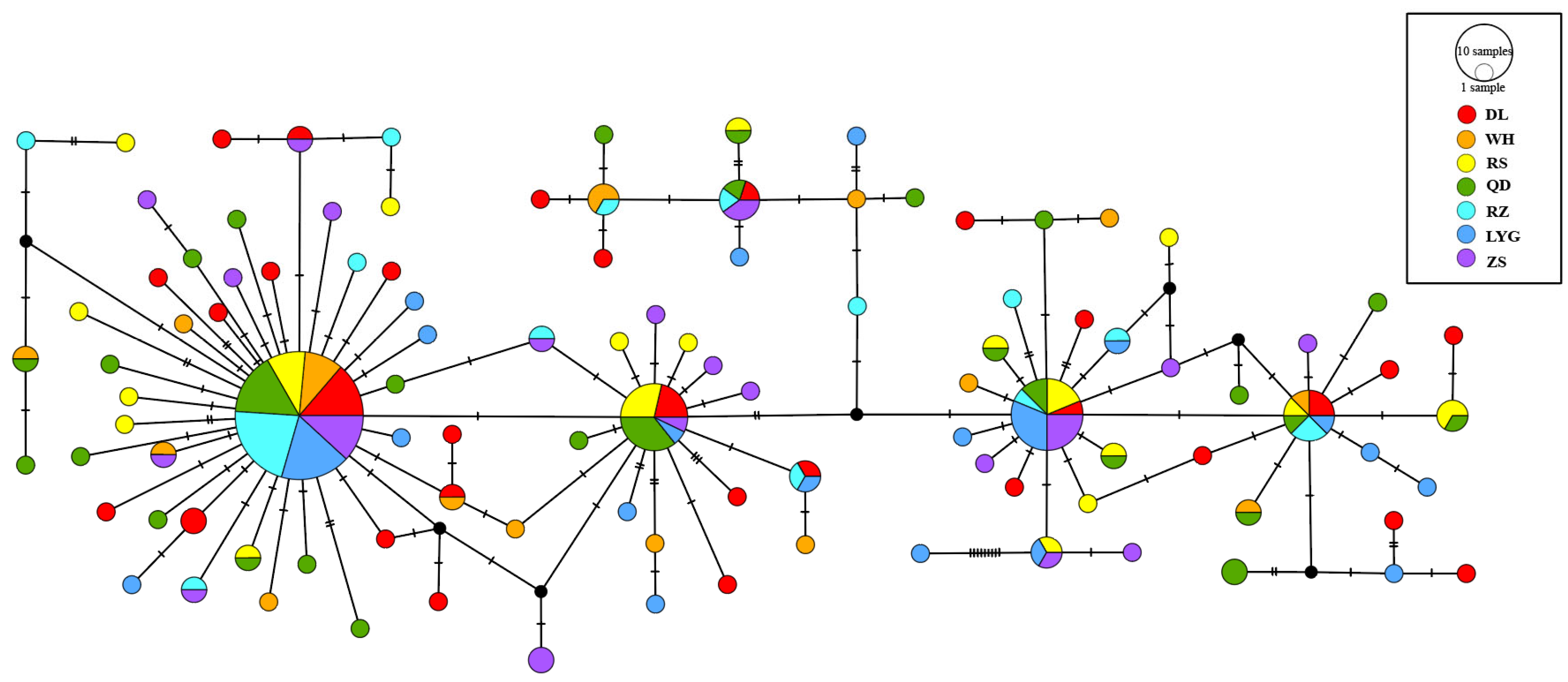
3.2. Haplotype Distribution and Genetic Diversity
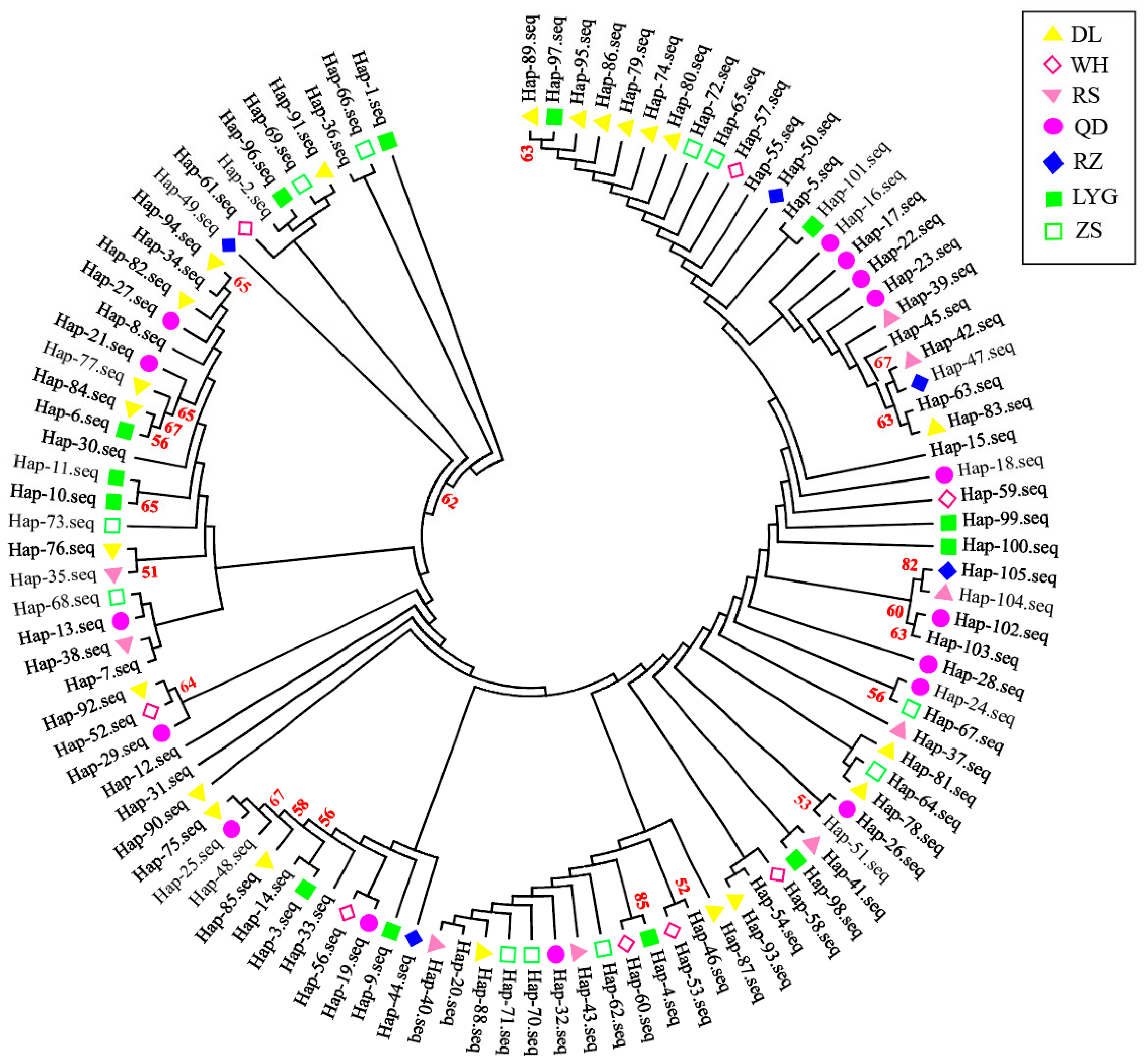
3.3. Genetic Distance and Population Differentiation
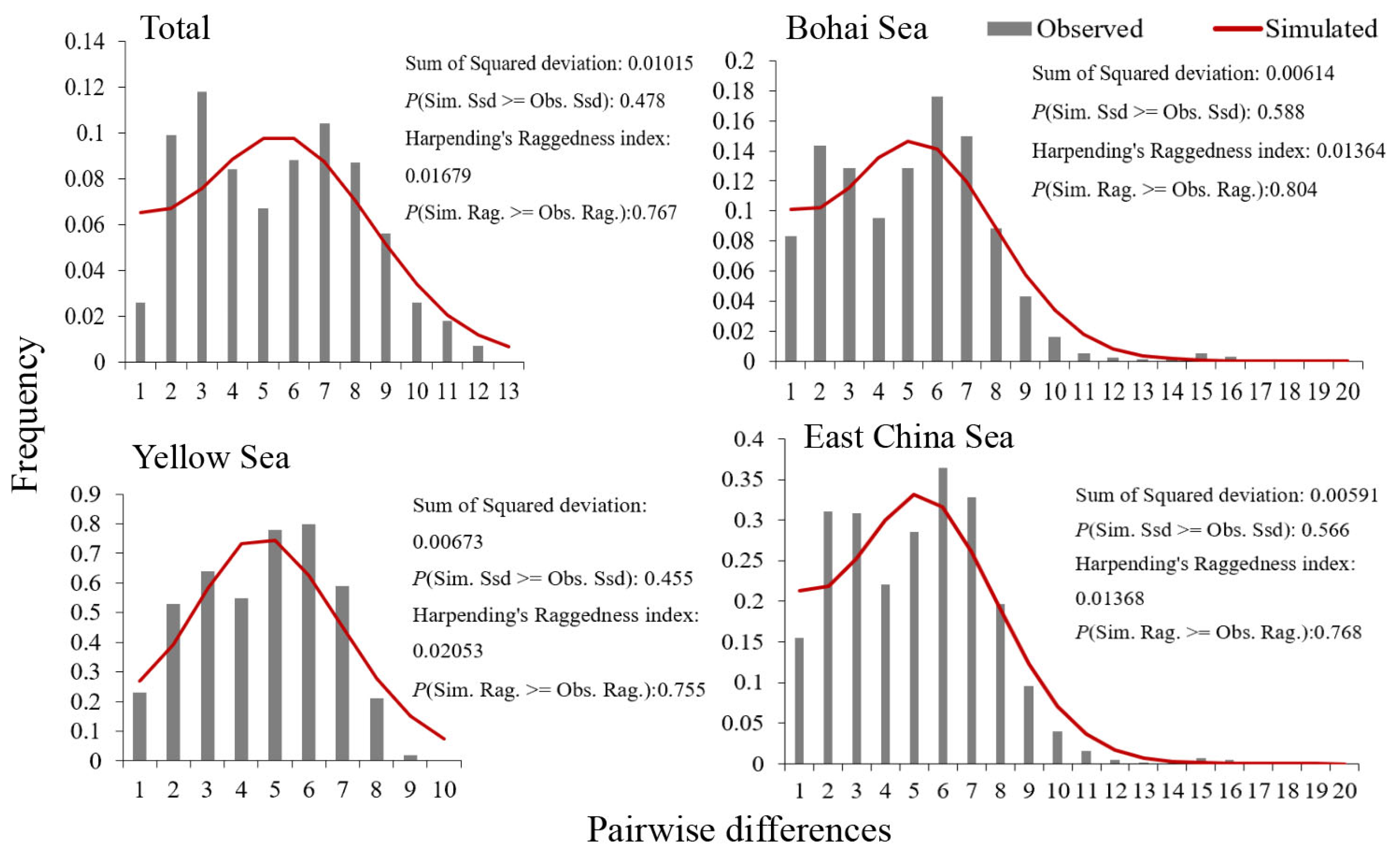
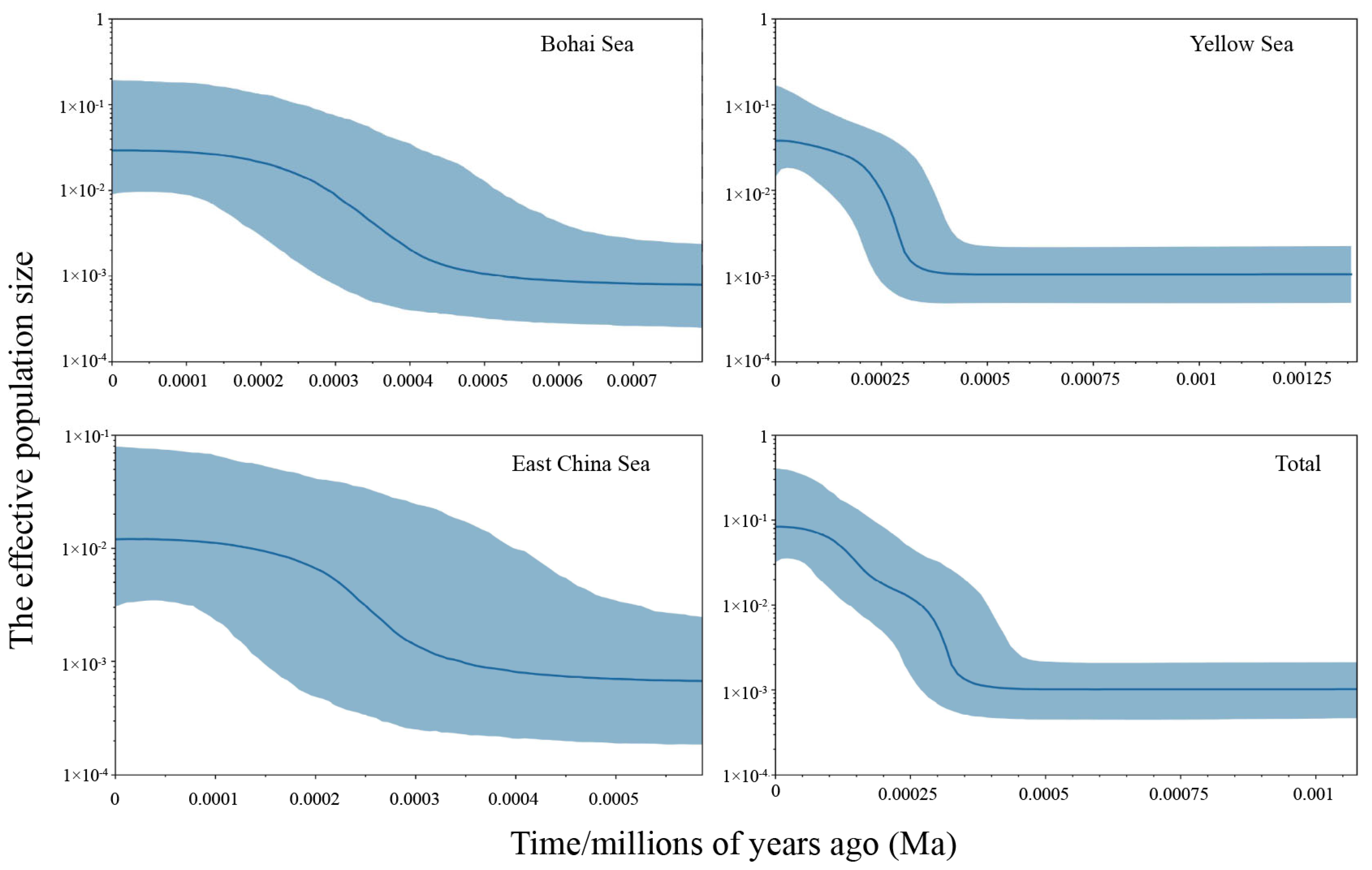
3.4. Demographic History
4. Discussion
4.1. Genetic Diversity and Variation
4.2. Genetic Structure and Population Homogeneity
4.3. Historical Population Dynamics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brevoort, J.C. Notes on Some Figures of Japanese Fish Taken from Recent Specimens by the Artists of the U. S. Japan Expedition; FAO: Washington, DC, USA, 1856; pp. 253–288. [Google Scholar]
- Chen, D.G.; Zhang, M.Z. Marine Fishes of China; China Ocean University Press: Qingdao, China, 2015; pp. 187–188. (In Chinese) [Google Scholar]
- Kimura, Y.; Ishikawa, S.; Tokai, T.; Nishida, M.; Tsukamoto, K. Early life history characteristics and genetic homogeneity of Conger myriaster leptocephali along the east coast of central Japan. Fish. Res. 2004, 70, 61–69. [Google Scholar] [CrossRef]
- Mu, X.X.; Zhang, C.; Zhang, C.L.; Yang, J.; Ren, Y.P. Age-structured otolith chemistry profiles revealing the migration of Conger myriaster in China Seas. Fish. Res. 2021, 239, 105938. [Google Scholar] [CrossRef]
- Tang, Y.L.; Sun, G.W.; Zhao, T.Y.; Liang, Z.L.; Yang, B.Z.; Chen, B. Mesh size selectivity of Conger myriaster pot. J. Fish. Sci. China 2010, 17, 136–142. (In Chinese) [Google Scholar]
- Zhang, Y.M.; Zhang, S.H.; Zhuang, Q.H.; Song, W.Y. Factory farming technology of Conger myriaster. Breed. Feed 2020, 19, 38–39. (In Chinese) [Google Scholar]
- Katayama, S.; Ishida, T.; Shimizu, Y.; Yamanobe, A. Seasonal change in distribution of conger eel Conger myriaster off the Pacific coast south of Tohoku, north-eastern Japan. Fish. Sci. 2004, 70, 1–6. [Google Scholar] [CrossRef]
- Yang, H.; Shi, B.; Niu, H.X.; Zhang, D.Q.; Li, J. Advances and future prospects in Conger myriaster research. Mar. Sci. 2020, 44, 152–158. (In Chinese) [Google Scholar]
- Ennos, R.A.; Worrell, R.; Arkle, P.; Malcolm, D.C. Genetic diversity and conservation. In Genetic Variation and Conservation of British Native Trees and Shrubs Current Knowledge and Policy Implications; Forestry Commission Technical: Edinburgh, UK, 2000; pp. 1–8. [Google Scholar]
- Nonić, M.; Šijačić-Nikolić, M. Genetic diversity: Sources, threats, and conservation. In Life on Land. Encyclopedia of the UN Sustainable Development Goals; Leal Filho, W., Azul, A.M., Brandli, L., Lange Salvia, A., Wall, T., Eds.; Springer: Cham, Switzerland, 2021; pp. 1–15. [Google Scholar] [CrossRef]
- Hughes, R.A.; Brian, D.; Inouye, B.D.; Marc, T.J.J.; Underwood, N.; Vellend, M. Ecological consequences of genetic diversity. Ecol. Lett. 2008, 11, 609–623. [Google Scholar] [CrossRef]
- Pinsky, M.L.; Palumbi, S.R. Meta-analysis reveals lower genetic diversity in overfished populations. Mol. Ecol. 2014, 23, 29–39. [Google Scholar] [CrossRef]
- Truelove, N.K.; Box, S.J.; Aiken, K.A.; Blythe-Mallett, A.; Boman, E.M.; Booker, C.J.; Byfield, T.T.; Cox, C.E.; Davis, M.; Delgado, G.A.; et al. Isolation by oceanic distance and spatial genetic structure in an overharvested international fishery. Divers. Distrib. 2017, 23, 1292–1300. [Google Scholar] [CrossRef]
- Melis, R.; Vacca, L.; Cuccu, D.; Mereu, M.; Cau, A.; Follesa, M.C.; Cannas, R. Genetic population structure and phylogeny of the common octopus Octopus vulgaris Cuvier, 1797 in the western Mediterranean Sea through nuclear and mitochondrial markers. Hydrobiologia 2018, 807, 277–296. [Google Scholar] [CrossRef]
- Madduppa, H.H.; Timm, J.; Kochzius, M. Reduced genetic diversity in the clown anemonefish Amphiprion ocellaris in exploited reefs of Spermonde Archipelago, Indonesia. Front. Mar. Sci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Kawazu, M.; Kameda, T.; Kurogi, H.; Yoda, M.; Ohshimo, S.; Sakai, T.; Tsukamoto, Y.; Mochioka, N. Biological characteristics of Conger myriaster during the initial stage of spawning migration in the East China Sea. Fish. Sci. 2015, 81, 663–671. [Google Scholar] [CrossRef]
- Wang, G.X.; Li, K.; Liu, R.C.; Jiang, Z.X.; Ba, X.B.; Song, Z.C.; Liu, L.P. Analysis of lipid metabolism in main tissues of artificially induced ovarian development tissues of Conger myriaster. J. Fish. China 2024, 48, 91–101. (In Chinese) [Google Scholar]
- Shi, B.; Wang, C.G.; Tang, X.H.; Zhao, X.Y.; Yan, K.W.; Ma, X.D. A study on sperm ultrastructure in the whitespotted conger, Conger myriaster. Mar. Sci. 2023, 47, 112–120. (In Chinese) [Google Scholar]
- Li, Z.; Liu, R.; Liu, J.; Jiang, Z.; Ba, X.; Li, K.; Liu, L. Effects of flowing water stimulation on hormone regulation during the maturation process of Conger myriaster ovaries. Front. Physiol. 2024, 15, 1404834. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Wang, Y.; Huang, X.H.; Zheng, J.; Qi, L.B.; Fu, B.S.; Qin, L. Nutritional value of different parts from sea eel (Astroconger myriaster) determined by untargeted-lipidomic approach. Food Res. Int. 2023, 173, 113402. [Google Scholar] [CrossRef]
- Zhao, X.; Shi, B.; Wang, C.; Cheng, H.; Wang, C.; Ma, X.; Yan, K.; Chen, Y. Effects of dietary supplementation of compound probiotics on growth, non-specific immunity, intestinal digestive enzyme, and microbiota of whitespotted conger (Conger myriaster) in recirculating aquaculture system. Aquacult. Int. 2024, 32, 2713–2738. [Google Scholar] [CrossRef]
- Xiao, P.Y.; Jiang, X.Z.; Yang, L.; Zhao, Y.; Yang, T.Y. Multiple analysis for morphometric traits on body mass of different Conger myriaster populations in the Yellow Sea and the Bohai Sea of China. J. Ecol. 2024, 44, 131–138. (In Chinese) [Google Scholar]
- Chen, Y.; Yan, K.W.; Shi, B.; Wang, C.G.; Zhao, X.Y.; Ma, X.D. Morphometric analysis and influence of important morphological traits on the body weight and net body weight of Conger myriaster in an industrialized culture model. Prog. Fish. Sci. 2024, 45, 83–96. (In Chinese) [Google Scholar]
- Xiao, P.; Yang, T. Population morphometry of Conger myriaster (Anguilliformes: Congridae) along the coast of China: Implications for population structure and fishery management. Animals 2024, 14, 2007. [Google Scholar] [CrossRef]
- Nakamura, O.; Watanabe, T.; Kamiya, H.; Muramoto, K. Galectin containing cells in the skin and mucosal tissues in Japanese conger eel, Conger myriaster: An immunohistochemical study. Dev. Comp. Immunol. 2001, 25, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Kimura, Y.; Tokai, T.; Tsukamoto, K.; Nishida, M. Genetic variation in the mitochondrial and nuclear DNA of the Japanese conger Conger myriaster. Fish. Sci. 2001, 67, 1081–1087. [Google Scholar] [CrossRef]
- Zou, C.; Wang, L.; Kong, L.; Wang, Y.; Wu, Z.; Xu, J.; Song, A.; Liu, H.; You, F. High levels of genetic diversity and connectivity of whitespotted conger Conger myriaster in the East China Coast. Mar. Biodivers. 2020, 50, 47. [Google Scholar] [CrossRef]
- Yin, J.; Mu, X.X.; Zhang, C.L.; Xu, B.D.; Xue, Y.; Ren, Y.P. Comparison of morphological and genetic characteristics of Conger myriaster populations in China. J. Fish. China 2020, 44, 358–367. (In Chinese) [Google Scholar]
- McCosker, J.; Smith, D.G.; Tighe, K. Conger myriaster. The IUCN Red List of Threatened Species 2019, e.T199034A2552131. Available online: https://dx.doi.org/10.2305/IUCN.UK.2019-1.RLTS.T199034A2552131.en (accessed on 17 August 2024).
- Vieira, A.R.; Rodrigues, A.S.B.; Sequeira, V.; Neves, A.; Paiva, R.B.; Paulo, O.S.; Gordo, L.S. Genetic and morphological variation of the forkbeard, Phycis phycis (Pisces, Phycidae): Evidence of panmixia and recent population expansion along its distribution area. PLoS ONE 2016, 11, e0167045. [Google Scholar] [CrossRef]
- Porrini, L.P.; Iriarte, P.J.F.; Iudica, C.M.; Abud, E.A. Population genetic structure and body shape assessment of Pagrus pagrus (Linnaeus, 1758) (Perciformes: Sparidae) from the Buenos Aires coast of the Argentine Sea. Neotrop. Ichthyol. 2015, 13, 431–438. [Google Scholar] [CrossRef]
- Zardoya, R.; Meyer, A. The complete nucleotide sequence of the mitochondrial genome of the lungfish (Protopterus dolloi) supports its phylogenetic position as a close relative of land vertebrates. Genetics 1996, 142, 1249–1263. [Google Scholar] [CrossRef]
- Maniatis, T.; Fritsch, E.F.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: New York, NY, USA, 1982; pp. 129–178. [Google Scholar]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Complete mitochondrial DNA sequence of Conger myriaster (Teleostei: Anguilliformes): Novel gene order for vertebrate mitochondrial genomes and the phylogenetic implications for anguilliform families. J. Mol. Evol. 2001, 52, 311–320. [Google Scholar] [CrossRef]
- Burland, T.G. Dnastar’s Lasergene sequence analysis software. In Bioinformatics Methods and Protocols. Methods in Molecular Biology™; Misener, S., Krawetz, S.A., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 132, pp. 71–91. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; pp. 10–88. [Google Scholar]
- Lynch, M.; Crease, T.J. The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 1990, 7, 377–394. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 3, 783–791. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. An exact test for population differentiation. Evolution 1995, 49, 1280–1283. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Luo, J.Y.; Xiong, F.; Liu, H.Y.; Zhai, D.D. Genetic structure of Pseudobrama simoni populations in the mainstream and tributaries of three Gorges Reservoir. J. Hydroecol. 2023, 44, 124–131. (In Chinese) [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, E.S.; McCafferty, A. Molecular Systematics of Fishes; Academic Press: New York, NY, USA, 1997; pp. 113–126. [Google Scholar]
- Ji, Z.W.; Su, B. Principles and Methodologies of Genetic Diversity Studies; Zhejiang Science and Technology Publishing House: Hangzhou, China, 1999; pp. 1–2. (In Chinese) [Google Scholar]
- Grant, W.A.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Ojango, J.M.; Mpofu, N.; Marshall, K.; Andersson-Eklund, L. Quantitative methods to improve the understanding and utilisation of animal genetic resources. In Animal Genetics Training Resource, Version 3; Ojango, J.M., Malmfors, B., Okeyo, A.M., Eds.; International Livestock Research Institute: Nairobi, Kenya; Swedish University of Agricultural Sciences: Uppsala, Sweden, 2011. [Google Scholar]
- Bian, G.M.; Wang, N.L.; Hu, Z.H.; Wang, Y.B.; Hu, C.S.; Chai, X.J. A comparative analysis on the genetic diversity of Trichiurus lepturus. Acta Hydrobiol. Sin. 2019, 43, 282–290. (In Chinese) [Google Scholar]
- Guo, D.D.; Liu, F.; Niu, B.L.; Lou, B. Genetic diversity of wild and cultured populations of little yellow croaker (Larimichthys polyactis) based on mitochondrial Cytb gene and D-loop region. Acta Agricul. Zhejiangensis 2022, 34, 1856–1865. (In Chinese) [Google Scholar]
- Peng, M.; Xiao, S.; Yao, J.X.; Pan, C.Y.; Li, M.; Gao, X.M.; Feng, P.F.; Li, M.Y.; Zeng, D.G.; Zhu, W.L.; et al. Genetic structure of four Sillago sihama populations in southern China based on mitochondrial sequences. J. Hydroecol. 2023, 44, 108–116. (In Chinese) [Google Scholar]
- Meyer, A. Evolution of mitochondrial DNA in Fishes. In Biochemistry and Molecular Biology of Fishes; Hochachka, P.W., Mommsen, T.P., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; Volume 2, pp. 1–38. [Google Scholar]
- Laura, M.; Ling, S.; Lee, Y.B.; Eva, G.V. Population genetic structure of the European conger (Conger conger) in North East Atlantic and West Mediterranean Sea. Fish. Res. 2016, 174, 245–249. [Google Scholar] [CrossRef]
- Correia, A.T.; Ramos, A.A.; Barros, F.; Silva, G.; Hamer, P.; Morais, P.; Cunha, R.L.; Castilho, R. Population structure and connectivity of the European conger eel (Conger conger) across the north-eastern Atlantic and western Mediterranean: Integrating molecular and otolith elemental approaches. Mar. Biol. 2012, 159, 1509–1525. [Google Scholar] [CrossRef]
- Billington, N.; Hebert, P.B.N. Mitochondria1 DNA diversity in fishes and its implications for introductions. Can. J. Fish. Aquat. Sci. 1991, 48, 88–94. [Google Scholar] [CrossRef]
- Yang, X.G.; Wang, Y.Q.; Zhou, K.Y.; Liu, Z.Q. Authentication of Oviductus ranae and its original animals using molecular marker. Biol. Pharm. Bull. 2002, 25, 1035–1039. [Google Scholar] [CrossRef]
- Holsinger, K.; Weir, B. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Variability within and among natural populations. In Evolution and the Genetics of Populations; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4, pp. 253–256. [Google Scholar]
- Balloux, F.; Lugon-Moulin, N. The estimation of population differentiation with microsatellite markers. Mol. Ecol. 2002, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Selkoe, K.A.; Watson, J.; Siegel, D.A.; Zacherl, D.C.; Toonen, R.J. Ocean currents help explain population genetic structure. Proceedings. Biol. Sci. 2010, 277, 1685–1694. [Google Scholar] [CrossRef]
- Gorie, S.; Tanda, M.; Nagasawa, K. Habitat preference and feeding habits of juvenile white-spotted conger Conger myriaster in the eastern Seto Inland Sea, Japan. Aquac. Sci. 2010, 58, 167–179. [Google Scholar] [CrossRef]
- Ulmo-Diaz, G.; Engman, A.; McLarney, W.O.; Lasso Alcalá, C.A.; Hendrickson, D.; Bezault, E.; Feunteun, E.; Prats-Léon, F.L.; Wiener, J.; Maxwell, R.; et al. Panmixia in the American eel extends to its tropical range of distribution: Biological implications and policymaking challenges. Evol. Appl. 2023, 16, 1872–1888. [Google Scholar] [CrossRef]
- Als, T.D.; Hansen, M.M.; Maes, G.E.; Castonguay, M.; Riemann, L.; Aarestrup, K.; Munk, P.; Sparholt, H.; Hanel, R.; Bernatchez, L. All roads lead to home: Panmixia of European eel in the Sargasso Sea. Mol. Ecol. 2011, 20, 1333–1346. [Google Scholar] [CrossRef]
- Zhai, D.D.; Wang, D.; He, T.Q.; Cai, F.T.; Duan, X.B.; Hu, X.K.; Zhu, B.; Xie, Z.G.; Xia, M.; Liu, H.Y.; et al. Genetic diversity and population historical dynamics of Ochetobus elongatus in the middle reaches of the Yangtze River. Acta Hydrobiol. Sin. 2024, 48, 1736–1744. (In Chinese) [Google Scholar]
- Kelletat, D.H. Pleistocene Epoch. In Encyclopedia of Coastal Science; Finkl, C.W., Makowski, C., Eds.; Encyclopedia of Earth Sciences Series; Springer: Cham, Switzerland, 2019; pp. 1379–1381. [Google Scholar]
- Wang, Q.H.; Wu, R.X.; Li, Z.L.; Niu, S.F.; Zhai, Y.; Huang, M.; Li, B. Effects of late Pleistocene climatic fluctuations on the phylogeographic and demographic history of Japanese scad (Decapterus maruadsi). Front. Mar. Sci. 2022, 9, 878506. [Google Scholar] [CrossRef]
- Liu, J.X.; Gao, T.X.; Zhuang, Z.M.; Jin, X.S.; Yokogawa, K.; Zhang, Y.P. Late Pleistocene divergence and subsequent population expansion of two closely related fish species, Japanese anchovy (Engraulis japonicus) and Australian anchovy (Engraulis australis). Mol. Phylogenet. Evol. 2006, 40, 712–723. [Google Scholar] [CrossRef]
- Han, Z.Q.; Gao, T.X.; Yanagimoto, T.; Sakurai, Y. Deep phylogeographic break among white croaker Pennahia argentata (Sciaenidae, Perciformes) populations in North-western Pacific. Fish. Sci. 2008, 74, 770–780. [Google Scholar] [CrossRef]
- Liu, M. Population Genetics of Pacific Herring and Pacific Cod in North Pacific. Ph.D. Dissertation, Ocean University of China, Qingdao, China, 2010. (In Chinese). [Google Scholar]
- Zhang, Z.D.; Yang, J.S. A Brief History of Marine Fisheries in China; Ocean Press: Beijing, China, 1983; pp. 5–14. (In Chinese) [Google Scholar]
- Brian, F. Fishing: How the Sea Fed Civilization; Yale University Press: London, UK, 2017; pp. 216–290. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Sites | Sea Regions | Geographic Coordinates | Sampling Date | Sample Size | Total Length (TL) | |
|---|---|---|---|---|---|---|
| Range/cm | Mean ± SD/cm | |||||
| Dalian (DL) | Bohai Sea | 38°55′ N; 121°37′ E | May 2023 | 40 | 33.7–51.7 | 45.7 ± 3.9 |
| Weihai (WH) | Yellow Sea | 37°31′ N; 122°7′ E | September 2022 | 20 | 28.9–36.6 | 32.7 ± 2.2 |
| Rushan (RS) | 36°56′ N; 121°32′ E | October 2022 | 29 | 27.6–45.7 | 35.0 ± 4.3 | |
| Qingdao (QD) | 36°4′ N; 120°23′ E | September 2022 | 41 | 30.6–39.2 | 35.0 ± 2.1 | |
| Rizhao (RZ) | 35°25′ N; 119°32′ E | August 2022 | 25 | 33.9–48.4 | 38.2 ± 3.3 | |
| Lianyungang (LYG) | 34°36′ N; 119°13′ E | April 2023 | 32 | 30.6–50.4 | 39.1 ± 3.7 | |
| Zhoushan (ZS) | East China Sea | 29°59′ N; 122°12′ E | October 2022 | 30 | 30.6–45.1 | 40.8 ± 3.9 |
| Population | Sample Size (n) | Number of Polymorphic Sites (S) | Number of Haplotypes (h) | Haplotype Diversity (Hd) | Nucleotide Diversity (π) | Average Number of Nucleotide Differences (k) |
|---|---|---|---|---|---|---|
| DL | 40 | 39 | 30 | 0.9667 ± 0.019 | 0.00389 ± 0.0022 | 4.445 ± 2.2381 |
| WH | 20 | 22 | 15 | 0.9421 ± 0.043 | 0.00368 ± 0.0021 | 4.205 ± 2.1792 |
| RS | 29 | 29 | 19 | 0.9507 ± 0.024 | 0.00357 ± 0.0021 | 4.074 ± 2.0916 |
| QD | 41 | 34 | 28 | 0.9512 ± 0.023 | 0.00372 ± 0.0021 | 4.250 ± 2.1512 |
| RZ | 25 | 19 | 14 | 0.8133 ± 0.080 | 0.00279 ± 0.0017 | 3.180 ± 1.7012 |
| LYG | 32 | 35 | 20 | 0.9073 ± 0.041 | 0.00388 ± 0.0022 | 4.423 ± 2.2406 |
| ZS | 30 | 25 | 20 | 0.9471 ± 0.027 | 0.00317 ± 0.0018 | 3.621 ± 1.8881 |
| Total | 217 | 105 | 105 | 0.9338 ± 0.013 | 0.00354 ± 0.0020 | 4.047 ± 2.0281 |
| Population | DL | WH | RS | QD | RZ | LYG | ZS |
|---|---|---|---|---|---|---|---|
| DL | — | 0.01908 (0.90918) | −0.00067 (0.35254) | −0.01079 (0.82617) | −0.01630 (0.88379) | 0.00008 (0.35449) | −0.00660 (0.56055) |
| WH | 0.00373 | — | 0.00164 (0.32129) | −0.01906 (0.87793) | −0.02659 (0.92969) | 0.00151 (0.34570) | −0.01083 (0.56445) |
| RS | 0.00374 | 0.00364 | — | −0.01010 (0.68848) | 0.01114 (0.22949) | −0.01836 (0.98438) | −0.01551 (0.80566) |
| QD | 0.00378 | 0.00365 | 0.00362 | — | −0.00913 (0.59570) | −0.00632 (0.54395) | −0.01011 (0.67871) |
| RZ | 0.00330 | 0.00316 | 0.00323 | 0.00324 | — | 0.01335 (0.18848) | −0.00713 (0.48828) |
| LYG | 0.00390 | 0.00380 | 0.00367 | 0.00379 | 0.00339 | — | −0.01094 (0.65918) |
| ZS | 0.00353 | 0.00340 | 0.00333 | 0.00343 | 0.00297 | 0.00350 | — |
| Packet Type | Source of Variation | d.f. | Sum of Squares | Variance Component | Percentage of Variation | ΦST |
|---|---|---|---|---|---|---|
| Seven populations | Among populations | 6 | 9.3400 | −0.01642 Va | −0.80 | −0.00803 (p = 0.86208) |
| Within populations | 210 | 432.900 | 2.06143 Vb | 100.80 | ||
| Total | 216 | 442.240 | 2.04501 | |||
| Three groups | Among groups | 2 | 2.656 | −0.00513 Va | −0.25 | −0.00911 (p = 0.86079) |
| Among populations within groups | 4 | 6.684 | −0.01348 Vb | −0.66 | ||
| Within populations | 210 | 432.900 | 2.06143 Vc | 100.91 | ||
| Total | 216 | 442.240 | 2.04282 |
| Test | DL | WH | RS | QD | RZ | LYG | ZS | Total |
|---|---|---|---|---|---|---|---|---|
| Tajima’s D | −1.80828 | −1.23315 | −1.62576 | −1.61079 | −1.32344 | −1.77369 | −1.51744 | −2.37500 |
| Tajima’s D p-value | 0.019 | 0.101 | 0.045 | 0.034 | 0.077 | 0.016 | 0.036 | 0.000 |
| Fu’s Fs | −25.22147 | −7.80182 | −10.51011 | −21.73441 | −6.16888 | −10.09990 | −13.17388 | −25.55153 |
| Fu’s Fs p-value | 0.000 | 0.001 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, P.; Yang, T. Genetic Diversity and Population Structure of Commercial Eel Conger myriaster (Anguilliformes: Congridae) Along the Coasts of China Based on Complete Mitochondrial Cyt b Sequences. Fishes 2025, 10, 41. https://doi.org/10.3390/fishes10020041
Xiao P, Yang T. Genetic Diversity and Population Structure of Commercial Eel Conger myriaster (Anguilliformes: Congridae) Along the Coasts of China Based on Complete Mitochondrial Cyt b Sequences. Fishes. 2025; 10(2):41. https://doi.org/10.3390/fishes10020041
Chicago/Turabian StyleXiao, Peiyi, and Tianyan Yang. 2025. "Genetic Diversity and Population Structure of Commercial Eel Conger myriaster (Anguilliformes: Congridae) Along the Coasts of China Based on Complete Mitochondrial Cyt b Sequences" Fishes 10, no. 2: 41. https://doi.org/10.3390/fishes10020041
APA StyleXiao, P., & Yang, T. (2025). Genetic Diversity and Population Structure of Commercial Eel Conger myriaster (Anguilliformes: Congridae) Along the Coasts of China Based on Complete Mitochondrial Cyt b Sequences. Fishes, 10(2), 41. https://doi.org/10.3390/fishes10020041