


Click Reactions in Kinetic Target-Guided Synthesis: Progress in the Discovery of Inhibitors for Biological Targets

Abstract
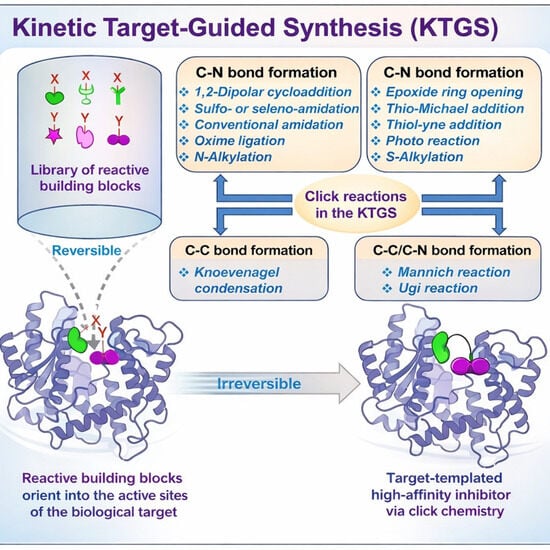
1. Introduction
2. Click Reactions in Kinetic Target-Guided Synthesis (KTGS)
2.1. Carbon-Nitrogen (C-N) Bond Forming Reactions
2.1.1. Alkyne-Azide Huisgen Cycloaddition (1,3-Dipolar Cycloaddition)
2.1.2. Sulfo- or Seleno-Click Amidation
2.1.3. Conventional Amidation
2.1.4. Oxime Ligation
2.1.5. N-Alkylation
2.2. C-S Bond Forming Reactions
2.2.1. S-Alkylation
2.2.2. Epoxide Ring Opening by Thiol
2.2.3. Thio-Michael Addition
2.2.4. Thiol-Yne Addition
2.2.5. Photo Reaction of Diazirine with Thiol
2.3. C-C Bond Formation
Knoevenagel Condensation
2.4. C-C and C-N Bond Formation
2.4.1. Mannich Reaction
2.4.2. Ugi Reaction
3. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TGS | Target-guided synthesis |
| DCC | Dynamic combinatorial chemistry |
| KTGS | Kinetic target-guided synthesis |
| AChE | Acetylcholinesterase |
| AChBPs | Acetylcholine binding proteins |
| nAChRs | Acetylcholine binding proteins |
| CAII | Carbonic anhydrase II |
| EthR | Ethionamide repressor |
| ABL | Abelson |
| BoNT/A | Botulinum neurotoxin serotype A |
| DAO | D-amino acid oxidase |
| BPL | Biotin protein ligase |
| IDE | Insulin-degrading enzyme |
| COX-2 | Cyclooxygenase-2 |
| HIV-1-Pr | HIV-1 protease |
| Bcl-xL | B-cell lymphoma-extra-large |
| Mcl-1 | Myeloid cell leukemia 1 |
| GAR TFase | Glycinamide ribonucleotide transformylase |
| FGAR | Formylglycinamide ribonucleotide |
| c-Src | Cellular Src kinase |
| EV protease | Enterovirus 3C-protease |
| ERAP2 | Endoplasmic reticulum aminopeptidase 2 |
| BSA | Bovine serum albumin |
| SmChi | Serratia marcescens chitinases |
| ITT | Insulin tolerance test |
| Fm | 9-Fluorenylmethyl |
| Glck | Glucokinase |
| pFLA | Pathogenic free-living amoebae |
| MAI | Multi-substrate adduct inhibitor |
References
- Sharpless, K.B.; Manetsch, R. In situ click chemistry: A powerful means for lead discovery. Expert Opin. Drug Discov. 2006, 1, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Hirsch, A.K.H. Dynamic combinatorial chemistry: A tool to facilitate the identification of inhibitors for protein targets. Chem. Soc. Rev. 2015, 44, 2455–2488. [Google Scholar] [CrossRef] [PubMed]
- Oueis, E.; Sabot, C.; Renard, P.Y. New insights into the kinetic target-guided synthesis of protein ligands. Chem. Commun. 2015, 51, 12158–12169. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.D.; Manetsch, R. Kinetic target-guided synthesis. Chem. Soc. Rev. 2010, 39, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Bosc, D.; Jakhlal, J.; Deprez, B.; Deprez-Poulain, R. Kinetic target-guided synthesis in drug discovery and chemical biology: A comprehensive facts and figures survey. Future Med. Chem. 2016, 8, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Jaegle, M.; Wong, E.L.; Tauber, C.; Nawrotzky, E.; Arkona, C.; Rademann, J. Protein-Templated Fragment Ligations-From Molecular Recognition to Drug Discovery. Angew. Chem. Int. Ed. 2017, 56, 7358–7378. [Google Scholar] [CrossRef] [PubMed]
- Bosc, D.; Camberlein, V.; Gealageas, R.; Castillo-Aguilera, O.; Deprez, B.; Deprez-Poulain, R. Kinetic Target-Guided Synthesis: Reaching the Age of Maturity. J. Med. Chem. 2020, 63, 3817–3833. [Google Scholar] [CrossRef] [PubMed]
- Unver, M.Y.; Gierse, R.M.; Ritchie, H.; Hirsch, A.K.H. Druggability Assessment of Targets Used in Kinetic Target-Guided Synthesis. J. Med. Chem. 2018, 61, 9395–9409. [Google Scholar] [CrossRef] [PubMed]
- Parvatkar, P.T.; Manetsch, R. The emergence of sulfo-click amidation in kinetic target-guided synthesis. Med. Chem. Res. 2024, 33, 1307–1314. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Wagner, A.; Manetsch, R. Biocompatible reactions: Advances in kinetic target-guided synthesis. Trends Chem. 2023, 5, 657–671. [Google Scholar] [CrossRef]
- Rideout, D. Self-Assembling Cytotoxins. Science 1986, 233, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Rideout, D.; Calogeropoulou, T.; Jaworski, J.; Mccarthy, M. Synergism through Direct Covalent Bonding between Agents–A Strategy for Rational Design of Chemotherapeutic Combinations. Biopolymers 1990, 29, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Huc, I.; Lehn, J.M. Virtual combinatorial libraries: Dynamic generation of molecular and supramolecular diversity by self-assembly. Proc. Natl. Acad. Sci. USA 1997, 94, 2016–2110. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- van der Vlag, R.; Yagiz Unver, M.; Felicetti, T.; Twarda-Clapa, A.; Kassim, F.; Ermis, C.; Neochoritis, C.G.; Musielak, B.; Labuzek, B.; Doemling, A.; et al. Optimized Inhibitors of MDM2 via an Attempted Protein-Templated Reductive Amination. ChemMedChem 2020, 15, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Gladysz, R.; Vrijdag, J.; Van Rompaey, D.; Lambeir, A.-M.; Augustyns, K.; De Winter, H.; Van der Veken, P. Efforts towards an on-target version of the Groebke-Blackburn-Bienayme (GBB) reaction for discovery of druglike urokinase (uPA) inhibitors. Chem.-Eur. J. 2019, 25, 12380–12393. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.M. Acetylcholinesterase: Enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Taylor, P.; Radic, Z. The cholinesterases: From genes to proteins. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 281–320. [Google Scholar] [CrossRef]
- Shafferman, A.; Barak, D.; Ordentlich, A.; Ariel, N.; Kronman, C.; Kaplan, D.; Velan, B. Structural and functional correlates of human acetylcholinesterase mutants for evaluating Alzheimer’s disease treatments: Functional analysis of E2020 and galanthamine HuAChE complexes. Adv. Behav. Biol. 2002, 51, 187–192. [Google Scholar]
- Changeux, J.-P.; Taly, A. Nicotinic receptors, allosteric proteins and medicine. Trends Mol. Med. 2008, 14, 93–102. [Google Scholar] [CrossRef]
- Role, L.; Kandel, E. Nicotinic Acetylcholine Receptors: From Molecular Biology to Cognition by Jean-Pierre Changeux and Stuart J. Edelstein. Neuron 2008, 58, 847–849. [Google Scholar] [CrossRef][Green Version]
- Changeux, J.-P. Nicotine addiction and nicotinic receptors: Lessons from genetically modified mice. Nat. Rev. Neurosci. 2010, 11, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- DeBarber, A.E.; Mdluli, K.; Bosman, M.; Bekker, L.-G.; Barry, C.E., III. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9677–9682. [Google Scholar] [CrossRef]
- Luttman, J.H.; Colemon, A.; Mayro, B.; Pendergast, A.M. Role of the ABL tyrosine kinases in the epithelial-mesenchymal transition and the metastatic cascade. Cell Commun. Signal. 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.J.; Tollervey, D. The function and synthesis of ribosomes. Nat. Rev. Mol. Cell Biol. 2001, 2, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Sacchi, S.; Murtas, G. Human D-Amino Acid Oxidase: Structure, Function, and Regulation. Front. Mol. Biosci. 2018, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.G.; Paparella, A.S.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Biotin Protein Ligase Is a Target for New Antibacterials. Antibiotics 2016, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.B. Aspartic proteinases in disease: A structural perspective. Curr. Drug Targets 2002, 3, 155–173. [Google Scholar] [CrossRef]
- Boot, R.G.; Blommaart, E.F.C.; Swart, E.; Ghuharali-Van der Vlugt, K.; Bijl, N.; Moe, C.; Place, A.; Aerts, J.M. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J. Biol. Chem. 2001, 276, 6770–6778. [Google Scholar] [CrossRef]
- Shahabuddin, M.; Toyoshima, T.; Aikawa, M.; Kaslow, D.C. Transmission-blocking activity of a chitinase inhibitor and activation of malarial parasite chitinase by mosquito protease. Proc. Natl. Acad. Sci. USA 1993, 90, 4266–4270. [Google Scholar] [CrossRef]
- Shibata, Y.; Foster, L.A.; Bradfield, J.F.; Myrvik, Q.N. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J. Immunol. 2000, 164, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.A.; Dixon, M.J.; Eggleston, I.M.; van Aalten, D.M.F. Natural product family 18 chitinase inhibitors. Nat. Prod. Rep. 2005, 22, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zheng, T.; Homer, R.J.; Kim, Y.-K.; Chen, N.Y.; Cohn, L.; Hamid, Q.; Elias, J.A. Acidic Mammalian Chitinase in Asthmatic Th2 Inflammation and IL-13 Pathway Activation. Science 2004, 304, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Hersh, L.B. The insulysin (insulin degrading enzyme) enigma. Cell. Mol. Life Sci. 2006, 63, 2432–2434. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Goto, S. Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef]
- Guo, Q.; Manolopoulou, M.; Bian, Y.; Schilling, A.B.; Tang, W.-J. Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-α, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol. 2010, 395, 430–443. [Google Scholar] [CrossRef]
- Authier, F.; Cameron, P.H.; Merlen, C.; Kouach, M.; Briand, G. Endosomal proteolysis of glucagon at neutral pH generates the bioactive degradation product miniglucagon-(19–29). Endocrinology 2003, 144, 5353–5364. [Google Scholar] [CrossRef]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes 2003, 52, 2315–2320. [Google Scholar] [CrossRef]
- Ciaccio, C.; Tundo, G.R.; Grasso, G.; Spoto, G.; Marasco, D.; Ruvo, M.; Gioia, M.; Rizzarelli, E.; Coletta, M. Somatostatin: A Novel Substrate and a Modulator of Insulin-Degrading Enzyme Activity. J. Mol. Biol. 2009, 385, 1556–1567. [Google Scholar] [CrossRef]
- Leissring, M.A.; Malito, E.; Hedouin, S.; Reinstatler, L.; Sahara, T.; Abdul-Hay, S.O.; Choudhry, S.; Maharvi, G.M.; Fauq, A.H.; Huzarska, M.; et al. Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS ONE 2010, 5, e10504. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.-J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Van der Donk, W.A.; Tsai, A.-L.; Kulmacz, R.J. The Cyclooxygenase Reaction Mechanism. Biochemistry 2002, 41, 15451–15458. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef]
- Marnett, L.J. The COXIB experience: A look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290. [Google Scholar] [CrossRef]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.F.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef]
- Walensky, L.D. BCL-2 in the crosshairs: Tipping the balance of life and death. Cell Death Differ. 2006, 13, 1339–1350. [Google Scholar] [CrossRef]
- San Chin, H.; Fu, N.Y. Physiological Functions of Mcl-1: Insights From Genetic Mouse Models. Front. Cell Dev. Biol. 2021, 9, 704547. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Kawamoto, H.; Honma, T.; Iwama, T.; Kim, S.-H. Structural basis for chemical inhibition of human blood coagulation factor Xa. Proc. Natl. Acad. Sci. USA 1998, 95, 6630–6635. [Google Scholar] [CrossRef]
- Inglese, J.; Blatchly, R.A.; Benkovic, S.J. A multisubstrate adduct inhibitor of a purine biosynthetic enzyme with a picomolar dissociation constant. J. Med. Chem. 1989, 32, 937–940. [Google Scholar] [CrossRef]
- Gardino, A.K.; Yaffe, M.B. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Biol. 2011, 22, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Goebel, K.; Eichler, S.; Wiendl, H.; Chavakis, T.; Kleinschnitz, C.; Meuth, S.G. The coagulation factors fibrinogen, thrombin, and factor xii in inflammatory disorders-a systematic review. Front. Immunol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Cumaraswamy, A.A.; Todic, A.; Resetca, D.; Minden, M.D.; Gunning, P.T. Inhibitors of Stat5 protein signalling. MedChemComm 2012, 3, 22–27. [Google Scholar] [CrossRef]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; De Palma, A.M.; Selisko, B.; Costenaro, L.; Papageorgiou, N.; Arnan, C.; Coutard, B.; Lantez, V.; De Lamballerie, X.; Baronti, C.; et al. Picornavirus non-structural proteins as targets for new anti-virals with broad activity. Antivir. Res. 2011, 89, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Raja, A.; Kuiper, J.J.W. Evolutionary immuno-genetics of endoplasmic reticulum aminopeptidase II (ERAP2). Genes Immun. 2023, 24, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.G.; Green, L.G.; Grynszpan, F.; Radic, Z.; Carlier, P.R.; Taylor, P.; Finn, M.G.; Sharpless, K.B. Click chemistry in situ: Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew. Chem. Int. Ed. 2002, 41, 1053–1057. [Google Scholar] [CrossRef]
- Radic, Z.; Taylor, P. Interaction kinetics of reversible inhibitors and substrates with acetylcholinesterase and its fasciculin 2 complex. J. Biol. Chem. 2001, 276, 4622–4633. [Google Scholar] [CrossRef]
- Carlier, P.R.; Han, Y.F.; Chow, E.S.H.; Li, C.P.L.; Wang, H.; Lieu, T.X.; Wong, H.S.; Pang, Y.-P. Evaluation of short-tether bis-THA acetylcholinesterase inhibitors. A further test of the dual binding site hypothesis. Bioorganic Med. Chem. 1999, 7, 351–357. [Google Scholar] [CrossRef]
- Taylor, P.; Lappi, S. Interaction of fluorescence probes with acetylcholinesterase. Site and specificity of propidium binding. Biochemistry 1975, 14, 1989–1997. [Google Scholar] [CrossRef]
- Manetsch, R.; Krasinski, A.; Radic, Z.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In Situ Click Chemistry: Enzyme Inhibitors Made to Their Own Specifications. J. Am. Chem. Soc. 2004, 126, 12809–12818. [Google Scholar] [CrossRef] [PubMed]
- Krasinski, A.; Radic, Z.; Manetsch, R.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In Situ Selection of Lead Compounds by Click Chemistry: Target-Guided Optimization of Acetylcholinesterase Inhibitors. J. Am. Chem. Soc. 2005, 127, 6686–6692. [Google Scholar] [CrossRef] [PubMed]
- Oueis, E.; Santoni, G.; Ronco, C.; Syzgantseva, O.; Tognetti, V.; Joubert, L.; Romieu, A.; Weik, M.; Jean, L.; Sabot, C.; et al. Reaction site-driven regioselective synthesis of AChE inhibitors. Org. Biomol. Chem. 2014, 12, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; El Achab, R.; Goerbig, D.M.; Morral, J.; Munoz-Torrero, D.; Badia, A.; Banos, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; et al. Synthesis, in Vitro Pharmacology, and Molecular Modeling of Very Potent Tacrine-Huperzine A Hybrids as Acetylcholinesterase Inhibitors of Potential Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 1999, 42, 3227–3242. [Google Scholar] [CrossRef]
- Camps, P.; El Achab, R.; Morral, J.; Munoz-Torrero, D.; Badia, A.; Banos, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; Luque, F.J. New tacrine-huperzine A hybrids (huprines): Highly potent tight-binding acetylcholinesterase inhibitors of interest for the treatment of Alzheimer’s disease. J. Med. Chem. 2000, 43, 4657–4666. [Google Scholar] [CrossRef]
- Grimster, N.P.; Stump, B.; Fotsing, J.R.; Weide, T.; Talley, T.T.; Yamauchi, J.G.; Nemecz, A.; Kim, C.; Ho, K.-Y.; Sharpless, K.B.; et al. Generation of Candidate Ligands for Nicotinic Acetylcholine Receptors via in situ Click Chemistry with a Soluble Acetylcholine Binding Protein Template. J. Am. Chem. Soc. 2012, 134, 6732–6740. [Google Scholar] [CrossRef]
- Horne, W.S.; Stout, C.D.; Ghadiri, M.R. A Heterocyclic Peptide Nanotube. J. Am. Chem. Soc. 2003, 125, 9372–9376. [Google Scholar] [CrossRef]
- Van Maarseveen, J.H.; Horne, W.S.; Ghadiri, M.R. Efficient Route to C2 Symmetric Heterocyclic Backbone Modified Cyclic Peptides. Org. Lett. 2005, 7, 4503–4506. [Google Scholar] [CrossRef]
- Angell, Y.L.; Burgess, K. Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. [Google Scholar] [CrossRef]
- Mocharla, V.P.; Colasson, B.; Lee, L.V.; Roeper, S.; Sharpless, K.B.; Wong, C.-H.; Kolb, H.C. In situ click chemistry: Enzyme-generated inhibitors of carbonic anhydrase II. Angew. Chem. Int. Ed. 2005, 44, 116–120. [Google Scholar] [CrossRef]
- Gao, J.; Cheng, X.; Chen, R.; Sigal, G.B.; Bruce, J.E.; Schwartz, B.L.; Hofstadler, S.A.; Anderson, G.A.; Smith, R.D.; Whitesides, G.M. Screening Derivatized Peptide Libraries for Tight Binding Inhibitors to Carbonic Anhydrase II by Electrospray Ionization-Mass Spectrometry. J. Med. Chem. 1996, 39, 1949–1955. [Google Scholar] [CrossRef]
- Grzybowski, B.A.; Ishchenko, A.V.; Kim, C.-Y.; Topalov, G.; Chapman, R.; Christianson, D.W.; Whitesides, G.M.; Shakhnovich, E.I. Combinatorial computational method gives new picomolar ligands for a known enzyme. Proc. Natl. Acad. Sci. USA 2002, 99, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Sigal, G.B.; Whitesides, G.M. Benzenesulfonamide-peptide conjugates as probes for secondary binding sites near the active site of carbonic anhydrase. Bioorganic Med. Chem. Lett. 1996, 6, 559–564. [Google Scholar] [CrossRef]
- Boriack, P.A.; Christianson, D.W.; Kingery-Wood, J.; Whitesides, G.M. Secondary Interactions Significantly Removed from the Sulfonamide Binding Pocket of Carbonic Anhydrase II Influence Inhibitor Binding Constants. J. Med. Chem. 1995, 38, 2286–2291. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Huang, S.G.; Whitesides, G.M. Lack of Effect of the Length of Oligoglycine- and Oligo(ethylene glycol)-Derived para-Substituents on the Affinity of Benzenesulfonamides for Carbonic Anhydrase II in Solution. J. Am. Chem. Soc. 1994, 116, 5057–5062. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, W.-Y.; Liu, K.; Lin, R.J.; Selke, M.; Kolb, H.C.; Zhang, N.; Zhao, X.-Z.; Phelps, M.E.; Shen, C.K.F.; et al. An integrated microfluidic device for large-scale in situ click chemistry screening. Lab Chip 2009, 9, 2281–2285. [Google Scholar] [CrossRef]
- Antti, H.; Sellstedt, M. Cell-Based Kinetic Target-Guided Synthesis of an Enzyme Inhibitor. ACS Med. Chem. Lett. 2018, 9, 351–353. [Google Scholar] [CrossRef]
- Willand, N.; Desroses, M.; Toto, P.; Dirie, B.; Lens, Z.; Villeret, V.; Rucktooa, P.; Locht, C.; Baulard, A.; Deprez, B. Exploring Drug Target Flexibility Using in Situ Click Chemistry: Application to a Mycobacterial Transcriptional Regulator. ACS Chem. Biol. 2010, 5, 1007–1013. [Google Scholar] [CrossRef]
- Peruzzotti, C.; Borrelli, S.; Ventura, M.; Pantano, R.; Fumagalli, G.; Christodoulou, M.S.; Monticelli, D.; Luzzani, M.; Fallacara, A.L.; Tintori, C.; et al. Probing the Binding Site of Abl Tyrosine Kinase Using in Situ Click Chemistry. ACS Med. Chem. Lett. 2013, 4, 274–277. [Google Scholar] [CrossRef][Green Version]
- Arioli, F.; Borrelli, S.; Colombo, F.; Falchi, F.; Filippi, I.; Crespan, E.; Naldini, A.; Scalia, G.; Silvani, A.; Maga, G.; et al. N-[2-Methyl-5-(triazol-1-yl)phenyl]pyrimidin-2-amine as a Scaffold for the Synthesis of Inhibitors of Bcr-Abl. ChemMedChem 2011, 6, 2009–2018. [Google Scholar] [CrossRef]
- Jin, X.; Daher, S.S.; Lee, M.; Buttaro, B.; Andrade, R.B. Ribosome-Templated Azide-Alkyne Cycloadditions Using Resistant Bacteria as Reaction Vessels: In Cellulo Click Chemistry. ACS Med. Chem. Lett. 2018, 9, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Glassford, I.; Teijaro, C.N.; Daher, S.S.; Weil, A.; Small, M.C.; Redhu, S.K.; Colussi, D.J.; Jacobson, M.A.; Childers, W.E.; Buttaro, B.; et al. Ribosome-Templated Azide-Alkyne Cycloadditions: Synthesis of Potent Macrolide Antibiotics by In Situ Click Chemistry. J. Am. Chem. Soc. 2016, 138, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Farrow, B.; Wong, M.; Malette, J.; Lai, B.; Deyle, K.M.; Das, S.; Nag, A.; Agnew, H.D.; Heath, J.R. Epitope Targeting of Tertiary Protein Structure Enables Target-Guided Synthesis of a Potent In-Cell Inhibitor of Botulinum Neurotoxin. Angew. Chem. Int. Ed. 2015, 54, 7114–7119. [Google Scholar] [CrossRef] [PubMed]
- Deyle, K.M.; Farrow, B.; Hee, Y.Q.; Work, J.; Wong, M.; Lai, B.; Umeda, A.; Millward, S.W.; Nag, A.; Das, S.; et al. A protein-targeting strategy used to develop a selective inhibitor of the E17K point mutation in the PH domain of Akt1. Nat. Chem. 2015, 7, 455–462. [Google Scholar] [CrossRef]
- Toguchi, S.; Hirose, T.; Yorita, K.; Fukui, K.; Barry, S.K.; Omura, S.; Sunazuka, T. In situ click chemistry for the identification of a potent D-amino acid oxidase inhibitor. Chem. Pharm. Bull. 2016, 64, 695–703. [Google Scholar] [CrossRef]
- Sparey, T.; Abeywickrema, P.; Almond, S.; Brandon, N.; Byrne, N.; Campbell, A.; Hutson, P.H.; Jacobson, M.; Jones, B.; Munshi, S.; et al. The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 3386–3391. [Google Scholar] [CrossRef]
- Duplantier, A.J.; Becker, S.L.; Bohanon, M.J.; Borzilleri, K.A.; Chrunyk, B.A.; Downs, J.T.; Hu, L.-Y.; El-Kattan, A.; James, L.C.; Liu, S.; et al. Discovery, SAR, and Pharmacokinetics of a Novel 3-Hydroxyquinolin-2(1H)-one Series of Potent D-Amino Acid Oxidase (DAAO) Inhibitors. J. Med. Chem. 2009, 52, 3576–3585. [Google Scholar] [CrossRef]
- Hondo, T.; Warizaya, M.; Niimi, T.; Namatame, I.; Yamaguchi, T.; Nakanishi, K.; Hamajima, T.; Harada, K.; Sakashita, H.; Matsumoto, Y.; et al. 4-Hydroxypyridazin-3(2H)-one Derivatives as Novel d-Amino Acid Oxidase Inhibitors. J. Med. Chem. 2013, 56, 3582–3592. [Google Scholar] [CrossRef]
- Hopkins, S.C.; Heffernan, M.L.R.; Saraswat, L.D.; Bowen, C.A.; Melnick, L.; Hardy, L.W.; Orsini, M.A.; Allen, M.S.; Koch, P.; Spear, K.L.; et al. Structural, Kinetic, and Pharmacodynamic Mechanisms of D-Amino Acid Oxidase Inhibition by Small Molecules. J. Med. Chem. 2013, 56, 3710–3724. [Google Scholar] [CrossRef]
- Kawazoe, T.; Tsuge, H.; Imagawa, T.; Aki, K.; Kuramitsu, S.; Fukui, K. Structural basis of D-DOPA oxidation by D-amino acid oxidase: Alternative pathway for dopamine biosynthesis. Biochem. Biophys. Res. Commun. 2007, 355, 385–391. [Google Scholar] [CrossRef]
- Tieu, W.; Soares da Costa, T.P.; Yap, M.Y.; Keeling, K.L.; Wilce, M.C.J.; Wallace, J.C.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Optimising in situ click chemistry: The screening and identification of biotin protein ligase inhibitors. Chem. Sci. 2013, 4, 3533–3537. [Google Scholar] [CrossRef]
- Soares da Costa, T.P.; Tieu, W.; Yap, M.Y.; Pendini, N.R.; Polyak, S.W.; Sejer Pedersen, D.; Morona, R.; Turnidge, J.D.; Wallace, J.C.; Wilce, M.C.J.; et al. Selective inhibition of Biotin Protein Ligase from Staphylococcus aureus. J. Biol. Chem. 2012, 287, 17823–17832. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Unver, M.Y.; Pal, A.; Bakker, M.; Berrier, S.P.; Hirsch, A.K.H. Fragment-Based Drug Design Facilitated by Protein-Templated Click Chemistry: Fragment Linking and -Optimization of Inhibitors of the Aspartic Protease Endothiapepsin. Chem.-Eur. J. 2016, 22, 14826–14830. [Google Scholar] [CrossRef] [PubMed]
- Koester, H.; Craan, T.; Brass, S.; Herhaus, C.; Zentgraf, M.; Neumann, L.; Heine, A.; Klebe, G. A Small Nonrule of 3 Compatible Fragment Library Provides High Hit Rate of Endothiapepsin Crystal Structures with Various Fragment Chemotypes. J. Med. Chem. 2011, 54, 7784–7796. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Sunazuka, T.; Sugawara, A.; Endo, A.; Iguchi, K.; Yamamoto, T.; Ui, H.; Shiomi, K.; Watanabe, T.; Sharpless, K.B.; et al. Chitinase inhibitors: Extraction of the active framework from natural argifin and use of in situ click chemistry. J. Antibiot. 2009, 62, 277–282. [Google Scholar] [CrossRef]
- Suzuki, K.; Sugawara, N.; Suzuki, M.; Uchiyama, T.; Katouno, F.; Nikaidou, N.; Watanabe, T. Chitinases A, B, and C1 of Serratia marcescens 2170 produced by recombinant Escherichia coli: Enzymatic properties and synergism on chitin degradation. Biosci. Biotechnol. Biochem. 2002, 66, 1075–1083. [Google Scholar] [CrossRef]
- Horn, S.J.; Soerlie, M.; Vaaje-Kolstad, G.; Norberg, A.L.; Synstad, B.; Vaarum, K.M.; Eijsink, V.G.H. Comparative studies of chitinases A, B and C from Serratia marcescens. Biocatal. Biotransform. 2006, 24, 39–53. [Google Scholar] [CrossRef]
- Rao, F.V.; Houston, D.R.; Boot, R.G.; Aerts, J.M.F.G.; Hodkinson, M.; Adams, D.J.; Shiomi, K.; Omura, S.; van Aalten, D.M.F. Specificity and Affinity of Natural Product Cyclopentapeptide Inhibitors against A. fumigatus, Human, and Bacterial Chitinases. Chem. Biol. 2005, 12, 65–76. [Google Scholar] [CrossRef]
- Houston, D.R.; Shiomi, K.; Arai, N.; Omura, S.; Peter, M.G.; Turberg, A.; Synstad, B.; Eijsink, V.G.H.; Van Aalten, D.M.F. High-resolution structures of a chitinase complexed with natural product cyclopentapeptide inhibitors: Mimicry of carbohydrate substrate. Proc. Natl. Acad. Sci. USA 2002, 99, 9127–9132. [Google Scholar] [CrossRef]
- Deprez-Poulain, R.; Hennuyer, N.; Bosc, D.; Liang, W.G.; Enee, E.; Marechal, X.; Charton, J.; Totobenazara, J.; Berte, G.; Jahklal, J.; et al. Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nat. Commun. 2015, 6, 8250. [Google Scholar] [CrossRef]
- Camberlein, V.; Fléau, C.; Sierocki, P.; Li, L.N.; Gealageas, R.; Bosc, D.; Guillaume, V.; Warenghem, S.; Leroux, F.; Rosell, M.; et al. Discovery of the First Selective Nanomolar Inhibitors of ERAP2 by Kinetic Target-Guided Synthesis. Angew. Chem. Int. Ed. 2022, 61, e202203560. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Whiting, M.; Muldoon, J.; Lin, Y.-C.; Silverman, S.M.; Lindstrom, W.; Olson, A.J.; Kolb, H.C.; Finn, M.G.; Sharpless, K.B.; Elder, J.H.; et al. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew. Chem. Int. Ed. 2006, 45, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Sun, J.; Wang, H.-G.; Manetsch, R. Bcl-XL-Templated Assembly of Its Own Protein-Protein Interaction Modulator from Fragments Decorated with Thio Acids and Sulfonyl Azides. J. Am. Chem. Soc. 2008, 130, 13820–13821. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Hu, X.; Doi, K.; Wang, H.-G.; Manetsch, R. Screening of Protein-Protein Interaction Modulators via Sulfo-Click Kinetic Target-Guided Synthesis. ACS Chem. Biol. 2011, 6, 724–732. [Google Scholar] [CrossRef]
- Namelikonda, N.K.; Manetsch, R. Sulfo-click reaction via in situ generated thioacids and its application in kinetic target-guided synthesis. Chem. Commun. 2012, 48, 1526–1528. [Google Scholar] [CrossRef]
- Nacheva, K.; Kulkarni, S.S.; Kassu, M.; Flanigan, D.; Monastyrskyi, A.; Iyamu, I.D.; Doi, K.; Barber, M.; Namelikonda, N.; Tipton, J.D.; et al. Going beyond Binary: Rapid Identification of Protein-Protein Interaction Modulators Using a Multifragment Kinetic Target-Guided Synthesis Approach. J. Med. Chem. 2023, 66, 5196–5207. [Google Scholar] [CrossRef] [PubMed]
- Kassu, M.; Parvatkar, P.T.; Milanes, J.; Monaghan, N.P.; Kim, C.; Dowgiallo, M.; Zhao, Y.Z.; Asakawa, A.H.; Huang, L.L.; Wagner, A.; et al. Shotgun Kinetic Target-Guided Synthesis Approach Enables the Discovery of Small-Molecule Inhibitors against Pathogenic Free-Living Amoeba Glucokinases. ACS Infect. Dis. 2023, 9, 2190–2201. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Parvatkar, P.T.; Wagner, A.; Kulkarni, S.S.; Manetsch, R. Chemoselective seleno-click amidation in kinetic target-guided synthesis. Chem. Commun. 2024, 60, 12722–12725. [Google Scholar] [CrossRef] [PubMed]
- Jaegle, M.; Steinmetzer, T.; Rademann, J. Protein-Templated Formation of an Inhibitor of the Blood Coagulation Factor Xa through a Background-Free Amidation Reaction. Angew. Chem. Int. Ed. 2017, 56, 3718–3722. [Google Scholar] [CrossRef]
- Schweinitz, A.; Stuerzebecher, A.; Stuerzebecher, U.; Schuster, O.; Stuerzebecher, J.; Steinmetzer, T. New substrate analogue inhibitors of factor Xa containing 4-amidinobenzylamide as P1 residue: Part 1. Med. Chem. 2006, 2, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Parvatkar, P.; Kato, N.; Uesugi, M.; Sato, S.-I.; Ohkanda, J. Intracellular Generation of a Diterpene-Peptide Conjugate that Inhibits 14-3-3-Mediated Interactions. J. Am. Chem. Soc. 2015, 137, 15624–15627. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Benkovic, S.J. Multisubstrate adduct inhibitors of glycinamide ribonucleotide transformylase: Synthetic and enzyme-assembled. Tetrahedron 1991, 47, 2351–2364. [Google Scholar] [CrossRef]
- Boger, D.L.; Haynes, N.E.; Warren, M.S.; Ramcharan, J.; Kitos, P.A.; Benkovic, S.J. Multisubstrate analogue based on 5,8,10-trideazafolate. Bioorganic Med. Chem. 1997, 5, 1853–1857. [Google Scholar] [CrossRef] [PubMed]
- Greasley, S.E.; Marsilje, T.H.; Cai, H.; Baker, S.; Benkovic, S.J.; Boger, D.L.; Wilson, I.A. Unexpected formation of an epoxide-derived multisubstrate adduct inhibitor on the active site of GAR transformylase. Biochemistry 2001, 40, 13538–13547. [Google Scholar] [CrossRef]
- Bigham, E.C.; Mallory, W.R.; Hodson, S.J.; Duch, D.S.; Ferone, R.; Smith, G.K. Multisubstrate analog inhibitors of glycinamide ribonucleotide transformylase based on 5-deazaacyclotetrahydrofolate (5-DACTHF). Heterocycles 1993, 35, 1289–1307. [Google Scholar] [CrossRef]
- Nguyen, R.; Huc, I. Using an enzyme’s active site to template inhibitors. Angew. Chem. Int. Ed. 2001, 40, 1774–1776. [Google Scholar] [CrossRef]
- Maki, T.; Kawamura, A.; Kato, N.; Ohkanda, J. Chemical ligation of epoxide-containing fusicoccins and peptide fragments guided by 14-3-3 protein. Mol. BioSyst. 2013, 9, 940–943. [Google Scholar] [CrossRef]
- Oueis, E.; Nachon, F.; Sabot, C.; Renard, P.-Y. First enzymatic hydrolysis/thio-Michael addition cascade route to synthesis of AChE inhibitors. Chem. Commun. 2014, 50, 2043–2045. [Google Scholar] [CrossRef]
- Kwarcinski, F.E.; Steffey, M.E.; Fox, C.C.; Soellner, M.B. Discovery of Bivalent Kinase Inhibitors via Enzyme-Templated Fragment Elaboration. ACS Med. Chem. Lett. 2015, 6, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Kwarcinski, F.E.; Fox, C.C.; Steffey, M.E.; Soellner, M.B. Irreversible Inhibitors of c-Src Kinase That Target a Nonconserved Cysteine. ACS Chem. Biol. 2012, 7, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Lossouarn, A.; Puteaux, C.; Bailly, L.; Tognetti, V.; Joubert, L.; Renard, P.Y.; Sabot, C. Metalloenzyme-Mediated Thiol-Yne Addition Towards Photoisomerizable Fluorescent Dyes. Chem.-Eur. J. 2022, 28, e202202180. [Google Scholar] [CrossRef] [PubMed]
- Puteaux, C.; Toubia, I.; Truong, L.; Hubert-Roux, M.; Bailly, L.; Oulyadi, H.; Renard, P.Y.; Sabot, C. Light-Induced Unlocking Reactivity of Fragments for Fast Target-Guided Synthesis of Carbonic Anhydrase Inhibitors. Angew. Chem. Int. Ed. 2024, 63, e202407888. [Google Scholar] [CrossRef] [PubMed]
- Tauber, C.; Wamser, R.; Arkona, C.; Tügend, M.; Aziz, U.B.; Pach, S.; Schulz, R.; Jochmans, D.; Wolber, G.; Neyts, J.; et al. Chemical Evolution of Antivirals Against Enterovirus D68 through Protein-Templated Knoevenagel Reactions. Angew. Chem. Int. Ed. 2021, 60, 13294–13301. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.L.; Nawrotzky, E.; Arkona, C.; Kim, B.G.; Beligny, S.; Wang, X.; Wagner, S.; Lisurek, M.; Carstanjen, D.; Rademann, J. The transcription factor STAT5 catalyzes Mannich ligation reactions yielding inhibitors of leukemic cell proliferation. Nat. Commun. 2019, 10, 66. [Google Scholar] [CrossRef]
- Weber, L. In vitro combinatorial chemistry to create drug candidates. Drug Discov. Today Technol. 2004, 1, 261–267. [Google Scholar] [CrossRef]
- Mancini, F.; Unver, M.Y.; Elgaher, W.A.M.; Jumde, V.R.; Alhayek, A.; Lukat, P.; Herrmann, J.; Witte, M.D.; Köck, M.; Blankenfeldt, W.; et al. Protein-Templated Hit Identification through an Ugi Four-Component Reaction. Chem.-Eur. J. 2020, 26, 14585–14593. [Google Scholar] [CrossRef] [PubMed]








































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Type of Click Reactions |
|---|---|
| 1 | C-N Bond Forming Reactions




 |
| 2 | C-S Bond Forming Reactions




 |
| 3 | C-C Bond Forming Reactions
 |
| 4 | C-C/C-N Bond Forming Reactions

 |
| Sr. No. | Biological Targets Used in KTGS and Their General Function | Type of Click Reactions Performed with the Target |
|---|---|---|
| 1 | Acetylcholinesterase (AChE) AChE is crucial for the hydrolysis of acetylcholine and other neurotransmitters within both the central and peripheral nervous systems [17,18]. Its inhibition is a therapeutic strategy employed in the management of Alzheimer’s disease, where AChE inhibitors serve to enhance cholinergic signaling and mitigate cognitive decline associated with the condition [19]. | 1,3-Dipolar cycloaddition, Thio-Michael addition |
| 2 | Acetylcholine binding proteins (AChBPs) Nicotinic acetylcholine receptors (nAChRs) are part of a larger superfamily of neurotransmitter ligand-gated ion channels. These receptors are recognized as promising therapeutic targets for various central nervous system disorders, including schizophrenia, nicotine dependence, and Alzheimer’s disease [20,21,22]. The acetylcholine-binding proteins (AChBPs) share structural homology with the extracellular domains of pentameric ligand-gated ion channels, providing valuable insights into the receptor’s function and pharmacology. | 1,3-Dipolar cycloaddition |
| 3 | Carbonic anhydrase II (CAII) CAII is a zinc-dependent metalloenzyme that plays crucial roles in several physiological processes, including cellular respiration and the regulation of CO2 and bicarbonate transport. They are also pivotal in acid-base homeostasis, bone resorption, and modulation of calcification. Additionally, CAII has been implicated in tumorigenesis, underscoring its importance in both normal physiological function and pathological states [23]. | 1,3-Dipolar cycloaddition, S-Alkylation, Thiol-yne addition, Photo reaction |
| 4 | Ethionamide repressor (EthR) EthR is a transcriptional regulator in mycobacteria that significantly influences the sensitivity of Mycobacterium tuberculosis to various antibiotics. When EthR is genetically inactivated, its levels increase, thereby enhancing the susceptibility of mycobacterial strains to the antibiotic ethionamide [24]. | 1,3-Dipolar cycloaddition |
| 5 | Abelson (ABL) The ABL family of tyrosine kinases, comprising ABL1 and ABL2, plays a critical role in regulating cellular processes essential for development and maintaining normal physiological conditions. However, during tumor progression, metastasis, tissue injury responses, inflammation, and neural degeneration, these ABL kinases can become inappropriately activated, contributing to the progression of various diseases [25]. | 1,3-Dipolar cycloaddition |
| 6 | Ribosome Ribosomes serve as the central components of the translation machinery in all organisms. The translation represents a crucial step in gene expression, as it converts the genetic information encoded in messenger RNAs (mRNAs) into continuous chains of amino acids (polypeptides or proteins) that possess structural and/or catalytic functions. Ribosomes perform the following two primary roles: decoding the genetic message and facilitating the formation of peptide bonds [26]. | 1,3-Dipolar cycloaddition |
| 7 | Botulinium neurotoxin serotype A (BoNT/A) BoNT/A is a zinc-dependent protease known for inhibiting the acetylcholine in response to calcium (Ca2+) signals. The structure of BoNT/A comprises the following two key components: a heavy chain that binds receptors and a light chain that harbors catalytic activity, linked by a disulfide bond. This mechanism enables BoNT/A to disrupt neurotransmission effectively [10]. | 1,3-Dipolar cycloaddition |
| 8 | D-amino acid oxidase (DAO) DAO is essential for catalyzing the oxidative deamination of D-amino acids, producing the corresponding α-keto acids, ammonia, and hydrogen peroxide. In humans, DAO is especially significant for its ability to convert D-serine, a neuromodulator involved in synaptic transmission and plasticity, into hydroxypyruvate via deamination. Proper regulation of D-serine levels by DAO is critical, as imbalances can have significant effects on neurological function and have been implicated in disorders such as schizophrenia and amyotrophic lateral sclerosis [27]. | 1,3-Dipolar cycloaddition |
| 9 | Biotin protein ligase (BPL) BPL is an essential enzyme found in all organisms, playing a crucial role in the post-translational attachment of biotin to a specific lysine residue located in the active site of biotin-dependent enzymes. Inhibitors targeting this vital metabolic enzyme, BPL, offer a promising avenue for developing new antibacterial drugs [28]. | 1,3-Dipolar cycloaddition |
| 10 | Aspartic protease endothiapsin Aspartic proteases constitute a diverse family of proteolytic enzymes distinguished by the presence of two highly conserved aspartic acid residues within their active sites, which are indispensable for catalytic activity. These enzymes are ubiquitously distributed across a broad spectrum of biological taxa, including fungi, vertebrates, plants, and retroviruses such as HIV. Their functional versatility allows them to participate in essential physiological and pathological processes, and their activity is closely linked to the development and progression of diseases such as hypertension, malaria, Alzheimer’s disease, and AIDS [29]. Endothiapepsin, a well-characterized fungal aspartic protease, is frequently employed as a structural and functional model in drug discovery owing to its significant homology with clinically relevant aspartic proteases. | 1,3-Dipolar cycloaddition, Ugi reaction |
| 11 | Chitinase Chitin is a constituent of fungal cell walls, the exoskeletons of crustaceans and insects, and the microfilarial sheaths of parasitic nematodes [30,31,32]. Chitinases have emerged as promising molecular targets for the development of antifungal, insecticidal, and antiparasitic agents, owing to their pivotal role in chitin degradation [33]. In addition to their significance in pathogen control, chitinases have garnered attention for their therapeutic potential in human medicine, particularly in the context of asthma and other chitin-associated inflammatory disorders [34]. | 1,3-Dipolar cycloaddition |
| 12 | Insulin-degrading enzyme (IDE) IDE is a zinc metalloprotease belonging to the M16 family, characterized by its broad substrate specificity and pivotal role in the catabolism of several physiologically significant peptides. IDE exhibits proteolytic activity towards insulin [35], amyloid-β [36], insulin-like growth factor II (IGF-II) [37], glucagon [38], amylin [39], and somatostatin [40], thereby regulating their extracellular and intracellular concentrations. This enzymatic activity emphasizes IDE’s significance in various metabolic processes and its potential implications in conditions such as diabetes and Alzheimer’s disease. | 1,3-Dipolar cycloaddition |
| 13 | Cyclooxygenase-2 (COX-2) Cyclooxygenase (COX) enzymes constitute a critical class of heme-containing isozymes that mediate the oxygenation of arachidonic acid to yield prostanoids. Dysregulation of COX activity has been implicated in the pathogenesis of a variety of pathological conditions, including inflammatory diseases, cardiovascular disorders, and certain cancers [41,42,43,44,45]. Consequently, COX enzymes represent key molecular targets for the development of anti-inflammatory and analgesic pharmacological agents. | 1,3-Dipolar cycloaddition |
| 14 | HIV-1 protease (HIV-1-Pr) HIV-1-Pr is a critical enzyme unique to the HIV-1 virus. It plays an essential role in the virus’s maturation by cleaving precursor polyproteins, specifically gag and gag-pol. This processing is vital for the correct assembly and replication of the retrovirus, which positions HIV-1-Pr as a key target for therapeutic strategies in the treatment of HIV/AIDS [46]. | 1,3-Dipolar cycloaddition |
| 15 | B-cell lymphoma-extra-large (Bcl-xL) Bcl-xL, a key member of the Bcl-2 protein family, is vital in regulating the intrinsic apoptosis pathway, the programmed cell death mechanism essential for maintaining cellular homeostasis and tissue development. Bcl-xL primarily acts as an anti-apoptotic factor by inhibiting the activation of pro-apoptotic proteins such as Bak and Bax. The balance between Bcl-xL and the pro-apoptotic members of the Bcl-2 family is crucial for determining cell survival and death. Dysregulation of Bcl-xL expression has been linked to the pathogenesis of various cancers and other diseases [47]. | Sulfo-click amidation |
| 16 | Myeloid cell leukemia 1 (Mcl-1) Mcl-1 is a multifaceted regulatory protein primarily recognized for its anti-apoptotic function within the Bcl-2 family. The Mcl-1 protein plays a crucial role in safeguarding cells from apoptosis under various conditions that induce cell death. Research has demonstrated that Mcl-1 is a vital pro-survival factor across multiple tumor types, leading to the recent development of Mcl-1-specific BH3-mimetics currently being evaluated in clinical trials [48]. | Sulfo-click amidation, Seleno-click amidation |
| 17 | Factor Xa Protein factor Xa is an essential serine protease in blood coagulation. It plays a crucial role in activating the coagulation cascade, making it a significant target for various antithrombotic medications to prevent blood clots [49]. | Conventional amidation |
| 18 | Glycinamide ribonucleotide transformylase (GAR TFase) GAR TFase is a folate-dependent enzyme that catalyzes a critical step in the de novo purine biosynthesis pathway, specifically mediating the transfer of a formyl group from 10-formyltetrahydrofolate to glycinamide ribonucleotide (GAR) to produce formylglycinamide ribonucleotide (FGAR). This reaction is essential for the generation of purine nucleotides, which are fundamental to nucleic acid synthesis and cellular proliferation. Due to its indispensable role in nucleotide biosynthesis, GAR TFase has long been recognized as a strategic molecular target for antineoplastic agents [50]. The primary role of GAR TFase is to formylate GAR. | N-Alkylation |
| 19 | 14-3-3 The 14-3-3 protein family comprises highly conserved, ubiquitously expressed dimeric proteins that function as key regulators of numerous intracellular signaling pathways, principally those mediated by serine/threonine kinases. The involvement of 14-3-3 proteins in these pathways underscores their role in maintaining cellular homeostasis and responding to diverse physiological stimuli. Their dysregulation has been implicated in several diseases, including cancer and neurodegenerative disorders, making them significant targets for therapeutic intervention [51]. | Epoxide ring formation Oxime ligation |
| 20 | Thrombin Thrombin is a member of the serine protease family, which converts fibrinogen into fibrin (an integral step in clot formation). Thrombin also participates in various other functions, including the initiation of inflammation, neoplastic transformation, angiogenesis, atherosclerosis, and tissue repair [52]. | Ugi reaction |
| 21 | Transcription factor STAT5 STAT5 is a transcription factor that becomes constitutively activated in various malignancies, where it regulates the expression of genes involved in cell proliferation, survival, and tumor progression [53]. Dysregulation or sustained activation of STAT5 has been linked to several malignancies, underscoring its potential as a therapeutic target. | Mannich reaction |
| 22 | Cellular Src kinase (c-Src) c-Src is an essential non-receptor tyrosine kinase that regulates various physiological processes and oncogenic activities. It is key to cellular signaling pathways, affecting normal cellular functions, and is associated with multiple cancer-related mechanisms. Its involvement in benign and malignant transformations underscores c-Src’s significance within the complex landscape of cell biology and cancer research [54]. | Thio-Michael addition |
| 23 | Enterovirus 3C-protease (EV protease) The Enterovirus 3C protease (EV protease) is crucial to the intricate dynamics of viral immune evasion. It is a well-established target for therapeutic interventions to treat enteroviral and rhinoviral infections. Studying the functions and mechanisms of the Enterovirus 3C protease provides valuable insights that can help develop effective strategies to combat viral infections [55]. | Knoevenagel condensation |
| 24 | Endoplasmic Reticulum Aminopeptidase 2 (ERAP2) ERAP2 is a zinc-dependent metallopeptidase localized within the lumen of the endoplasmic reticulum, where it plays a crucial role in antigen processing for presentation by the Class-I major histocompatibility complex. This process is essential for initiating immune responses against infected cells. Variations in the ERAP2 gene, particularly single-nucleotide polymorphisms, have been linked to increased susceptibility to chronic inflammatory disorders, such as Crohn’s disease, while also providing some protection against severe infections, such as pneumonia [56]. | 1,3-Dipolar cycloaddition |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Parvatkar, P.T.; Satam, N. Click Reactions in Kinetic Target-Guided Synthesis: Progress in the Discovery of Inhibitors for Biological Targets. Methods Protoc. 2026, 9, 54. https://doi.org/10.3390/mps9020054
Parvatkar PT, Satam N. Click Reactions in Kinetic Target-Guided Synthesis: Progress in the Discovery of Inhibitors for Biological Targets. Methods and Protocols. 2026; 9(2):54. https://doi.org/10.3390/mps9020054
Chicago/Turabian StyleParvatkar, Prakash T., and Nishikant Satam. 2026. "Click Reactions in Kinetic Target-Guided Synthesis: Progress in the Discovery of Inhibitors for Biological Targets" Methods and Protocols 9, no. 2: 54. https://doi.org/10.3390/mps9020054
APA StyleParvatkar, P. T., & Satam, N. (2026). Click Reactions in Kinetic Target-Guided Synthesis: Progress in the Discovery of Inhibitors for Biological Targets. Methods and Protocols, 9(2), 54. https://doi.org/10.3390/mps9020054