
Synthesis of DOTA-Based 43Sc Radiopharmaceuticals Using Cyclotron-Produced 43Sc as Exemplified by [43Sc]Sc-PSMA-617 for PSMA PET Imaging

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Experimental Design
2.1. Protocol Overview
2.2. Translation to Other Scandium Isotopes and DOTA-Based Radioligands
2.3. Materials
- Isotopically enriched [42Ca]CaCO3 (Isoflex USA, San Francisco, CA, USA)
- Trance analysis water (Honeywell, Charlotte, NC, USA; P/N 95305500ML or equivalent)
- HCl (trace metal grade) (Fisher Chemical, Pittsburgh, PA, USA; P/N A508-P500 or equivalent)
- HNO3 (trace metal grade) (Fisher Chemical, Pittsburgh, PA, USA; P/N A509P212 or equivalent)
- Acetic Acid (trace metal grade) (Fisher Chemical, Pittsburgh, PA, USA; P/N A507-P500 or equivalent)
- Ammonium Acetate (trace metal grade) (Sigma Aldrich, St. Louis, MO, USA; P/N 50-180-4364 or equivalent)
- Ammonium Formate (HPLC grade) (Honeywell, Charlotte, NC, USA; P/N 1784350G or equivalent)
- Methanol (HPLC Grade) (MilliporeSigma, Burlington, MA, USA; P/N MMX0475P6 or equivalent)
- Ethanol (200 proof)
- PSMA-617 (Vipivotide tetraxetan) (Selleckchem, Houston, TX, USA; P/N S8670)
- Phosphate-buffered saline (PBS)
- Ammonium oxalate monohydrate (trace-analysis grade) (Thermo Scientific Chemicals, Waltham, MA, USA; P/N 206275000 or equivalent)
- Ammonium Hydroxide (trace metal grade) (Fisher Chemical, Pittsburgh, PA, USA; P/N A512-P500 or equivalent)
- Non-Branched DGA Resin 2 mL Column (Eichrom, Lisle, IL, USA; P/N DN-R50-S)
- Oasis HLB light cartridge (30 mg sorbent) (Waters, Milford, MA, USA; P/N 186005125)
2.4. Equipment
- PTFE Coated Spatula (Fisher Scientific, Pittsburgh, PA, USA; P/N 13-820-058 or equivalent)
- Alumina crucible or combustion boat and cover (Fisher Scientific, Pittsburgh, PA, USA; P/N FB960I or equivalent)
- Box furnace (Lindberg/MPH, Riverside, MI, USA; BF51800 or equivalent)
- Pellet press and corresponding punch and die set (Parr Instrument Company, Moline, IL, USA; P/N 2810-2106-103101)
- Accumet AB15+ pH/mV/°C meter (Fisher Scientific, Pittsburgh, PA, USA; P/N 13-636-AB15PB or equivalent)
- Isotemp thermal mixer (Fisher Scientific, Pittsburgh, PA, USA; P/N 270600F or equivalent)
- Scan-Ram radioTLC scanner (LabLogic, Sheffield, UK)
- iTLC SG glass microfiber chromatography paper (Agilent Technologies, Santa Clara, CA, USA; P/N SGI0001)
- Infinity 1260 HPLC system (Agilent Technologies, Santa Clara, CA, USA; P/N 9572)
- Flow-Ram radioHPLC detector (LabLogic, Sheffield, UK)
- Wizard2 2480 gamma counter (Revvity, Waltham, MA, USA)
- Magnetic stirrer hot plate
- Microcentrifuge (compatible with 2 mL tubes)
- Fume hood with a nitrogen gas line
- Buchner funnel with inner joint
- Buchner funnel with filter disc
- 1000, 100, and 10 µL micropipettes
2.5. Laboratory Supplies
- Antistatic polystyrene weighing dishes (Fisher Scientific, Pittsburgh, PA, USA; P/N 08-732-112 or equivalent)
- 25 mL PFA beaker (Corning, Corning, NY, USA; P/N 1003P-25 or equivalent)
- 15 mL conical centrifuge tubes (Corning, Corning, NY, USA; P/N 352097 or equivalent)
- 2 mL microcentrifuge tubes (Fisher Scientific, Pittsburgh, PA, USA; P/N 05-408-138 or equivalent)
- Screw top septum vials (Fisher Scientific, Pittsburgh, PA, USA; P/N PI13019 or equivalent)
- Polystyrene tubes for Wizard2 2480 (Revvity, Waltham, MA, USA; P/N 50-905-2501)
- Whatman 42 quantitative ashless filter paper, GR42 (Cytiva, Marlborough, MA, USA; P/N 1442042)
- 1000, 100, and 10 µL filtered pipette tips
- Kimwipes
- Parafilm
3. Procedure
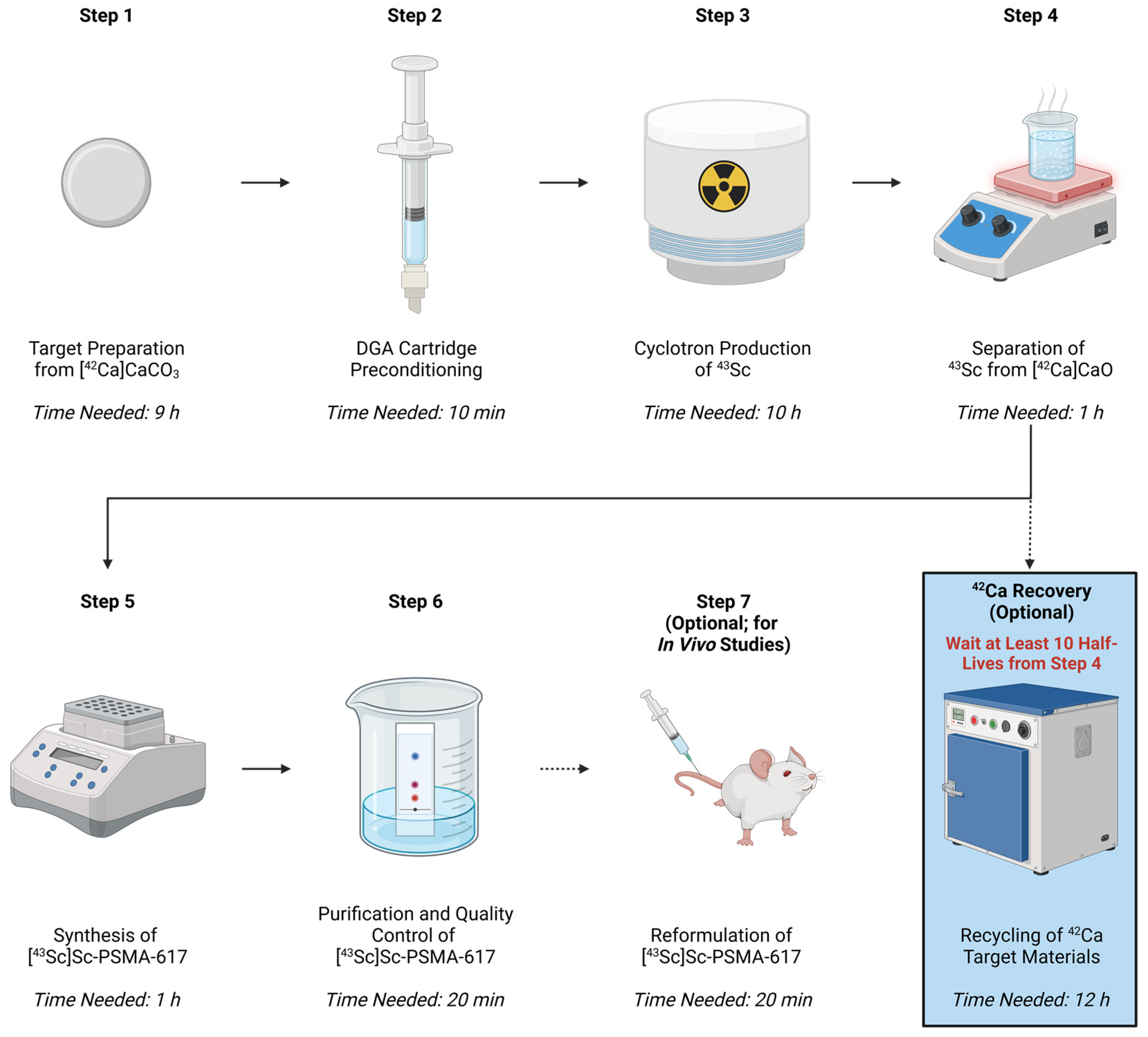
3.1. Synthesis of Injectable [43Sc]Sc-PSMA-617 from [42Ca]CaCO3—Total Time Needed: 22 h
3.1.1. Target Preparation from [42Ca]CaCO3—Time Needed: 9 h
 CRITICAL STEP The transfer of solid reagents described in this section (along with any transfers in subsequent sections) should only be performed with non-metal or PTFE coated tools. Failure to use non-metal or coated tools may result in metal contamination in the 43Sc solution. Metal contaminants will compete with 43Sc for the chelation complex in the radiolabeling portion of this procedure and may result in failed conjugation with PSMA-617 [46,48].
CRITICAL STEP The transfer of solid reagents described in this section (along with any transfers in subsequent sections) should only be performed with non-metal or PTFE coated tools. Failure to use non-metal or coated tools may result in metal contamination in the 43Sc solution. Metal contaminants will compete with 43Sc for the chelation complex in the radiolabeling portion of this procedure and may result in failed conjugation with PSMA-617 [46,48].- Transfer [42Ca]CaCO3 to an alumina crucible or combustion boat.
- When deciding the amount of [42Ca]CaCO3 to transfer, consider that calcination removes CO2 to produce [42Ca]CaO and thus the final target mass will be significantly lower than the mass of the starting material (1.0 mg of CaCO3 yields 0.56 mg of CaO).
- A quartz crucible can also be utilized in place of an alumina crucible; however, a quartz crucible will incur a significant buildup of static electricity, making it difficult to transfer the resultant [42Ca]CaO without loss.
- Place the [42Ca]CaCO3 in a laboratory furnace and gradually increase the furnace temperature to 800 °C in 100–150 °C increments.
- Maintain a temperature of 800 °C for 1 h to ensure complete conversion of [42Ca]CaCO3 to [42Ca]CaO.
- Shut off the furnace and allow it to return to room temperature (~ 8 h) before removing the crucible containing [42Ca]CaO.
- Scrape the [42Ca]CaO material from the crucible onto a piece of weighing paper. Carefully use the weighing paper to transfer the material into the die of the pellet press, as is shown in Figure 2.
![Mps 08 00058 i001]() CRITICAL STEP Ensure that the pellet press is free of residual material prior to use by wiping the die, die holder, and punch of the pellet press with an ethanol soaked Kimwipe. The presence of residual calcium on any of these parts can cause the resultant target to become brittle or malformed due to an uneven distribution of pressure during pressing
CRITICAL STEP Ensure that the pellet press is free of residual material prior to use by wiping the die, die holder, and punch of the pellet press with an ethanol soaked Kimwipe. The presence of residual calcium on any of these parts can cause the resultant target to become brittle or malformed due to an uneven distribution of pressure during pressing
- 6.
- Adjust the height of the die such that a non-negligible amount of force is required to fully press the punch into the bottom of the die.
![Mps 08 00058 i001]() CRITICAL STEP Improperly adjusting the die height will lead to broken targets (die is too high and thus too much pressure is exerted by the punch) and malformed targets (die is too low and not enough pressure is exerted by the punch).
CRITICAL STEP Improperly adjusting the die height will lead to broken targets (die is too high and thus too much pressure is exerted by the punch) and malformed targets (die is too low and not enough pressure is exerted by the punch).
- 7.
- Lower the punch into the die until the point that external force beyond gravity would be required to lower it any further. Gently rotate the die around the punch to evenly distribute the [42Ca]CaO material in the die.
- 8.
- Fully press the punch down into the die.
- 9.
- Lift the punch out of the die and retrieve the newly formed target.
CRITICAL STEP As the desired thickness of the target decreases, the structural integrity of the target decreases, making it substantially more difficult to maintain the desired disc shape of the target (which maximizes the target surface area in contact with the deuteron beam and thus maximizes yield). Therefore, if attempts to press a target are resulting in broken or imperfect discs, it is recommended to increase the mass of target material being pressed to reduce brittleness.- 10.
![Mps 08 00058 i002]() PAUSE STEP The calcified [42Ca]CaO target can be stored indefinitely in a parafilm wrapped weighing dish if it is kept under vacuum. This prevents the reabsorption of atmospheric carbon dioxide (reforming CaCO3) or water (forming Ca(OH)2) into the target. If the target has been exposed to atmospheric air for a prolonged period, the target can be re-calcinated into CaO by repeating the above procedure.
PAUSE STEP The calcified [42Ca]CaO target can be stored indefinitely in a parafilm wrapped weighing dish if it is kept under vacuum. This prevents the reabsorption of atmospheric carbon dioxide (reforming CaCO3) or water (forming Ca(OH)2) into the target. If the target has been exposed to atmospheric air for a prolonged period, the target can be re-calcinated into CaO by repeating the above procedure.

3.1.2. DGA Cartridge Preconditioning—Time Needed: 10 min
CRITICAL STEP Work described in this section should only be performed with metal-free or trace-analysis grade reagents. Failure to use such reagents may result in metal contamination in the 43Sc solution. Metal contaminants will compete with 43Sc for the chelation complex in the radiolabeling portion of this procedure and may result in failed conjugation with PSMA-617 [46,48]. CRITICAL STEP The preconditioning of the DGA cartridge described in this section is a procedure which must be executed at time-sensitive points relative to other portions of this protocol, notably the separation of 43Sc from the 42Ca target which begins in Section 3.1.4. Because it requires a slow gravity drip, the first phase of conditioning (Step 11) must be initiated approximately 24 h prior to the start of the separation of 43Sc from the 42Ca target. Failure to allow the DGA cartridge to gravity drip for enough time can lead to the cartridge being improperly conditioned, ultimately resulting in a failed trapping or elution of the desired [43Sc]ScCl3 product in Section 3.1.4. CRITICAL STEP The passing of any solution through the DGA column in Section 3.1.2. and Section 3.1.4. is done by loading the cartridge from the top and pushing the solution out from the bottom of the cartridge. Use of the reverse orientation (loading from the bottom and eluting from the top) may result in the unsuccessful trapping or elution of the 43Sc product.- 11.
- Draw 6 mL (three full-column volumes) of 2.5 M HNO3 into a needleless syringe. Connect the top of the DGA cartridge to the syringe and gently saturate the sorbent with HNO3. Vertically place the cartridge inside a conical tube so it can gravity drip for approximately 24 h.
- 12.
- Slowly push the remaining 2.5 M HNO3 through the cartridge at an approximate rate of 1.0 mL/s. Fill another needleless syringe with 6 mL (three ful- column volumes) of 5 M HNO3 and use it to gently wash the DGA column.
3.1.3. Cyclotron Production of 43Sc—Time Needed: 10 h
- 13.
- Transfer the [42Ca]CaO target into the cyclotron’s solid target holder and load it into the cyclotron.
- If using a retrofitted target holder, such as the beam stop-based target holder we describe in Meier et al., it is helpful to wrap the target in a thin graphite foil prior to transfer [50]. This does not impact any of the downstream separation chemistry and ensures that no target material is lost during loading or retrieval [50].
- 14.
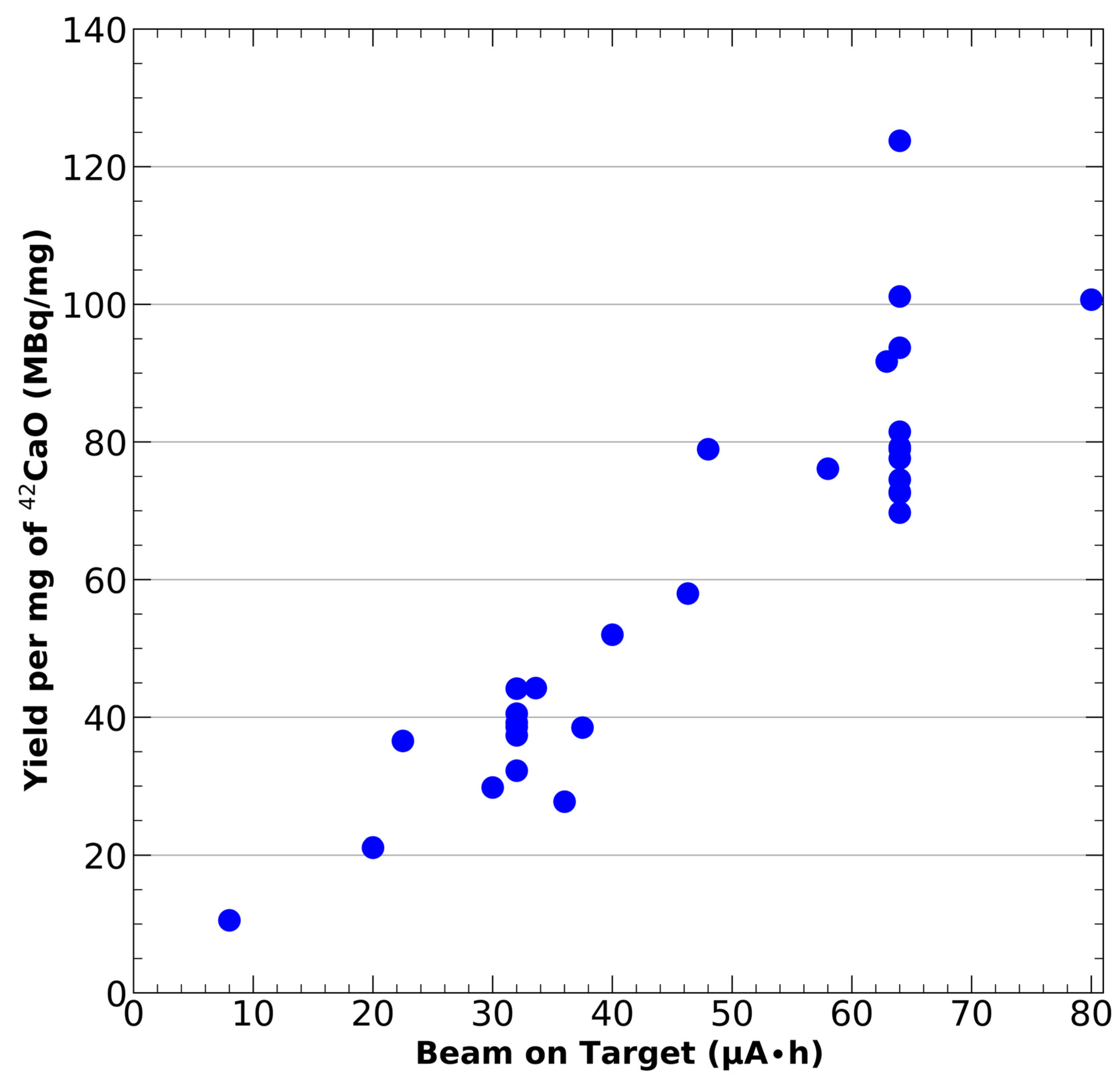
- Produce 43Sc via the 42Ca(d,n)43Sc reaction by irradiating the target with a deuteron beam. The deuteron current, irradiation time, and incident deuteron energy can all be adjusted based upon the desired activity amount and the cooling capabilities of the solid target holder. Increasing beam current and irradiation time will result in larger yields. The TENDL simulated 42Ca(d,n)43Sc cross-section suggests that the most optimal yields occur between 2–10 MeV [42]. For this publication, we have utilized a beam energy of 9 MeV, 2–10 μA of beam current, and an irradiation time of 8–10 h.
- 15.
- Approximately 1 h after the end of bombardment, retrieve the irradiated target from the cyclotron.
CRITICAL STEP It is essential that the target is kept inside the shielded cyclotron vault immediately after irradiation, so that the short-lived isotopes produced can decay prior to handling. While the abundance of these short-lived isotopes will vary with the isotopic composition of the target material and selected beam energy, potential short-lived contaminants originating from the target include: 46K (t1/2 = 96.31 s), 49Ca (t1/2 = 8.718 min), 44K (t1/2 = 22.13 min), and 49Sc (t1/2 = 57.20 min) [41,42,50]. If using a solid target holder retrofitted to the beam stop, such as that described in Meier et al., any titanium components in the target holder would also result in the production of short-lived 47V (32.6 min) [41,42,50]. Although not well characterized, as the production of 43Sc involves the ejection of a neutron from 42Ca, neutron activation of the cyclotron vault and aluminum in the target holder is also possible. With regards to the target holder this would result in the production of 28Al (t1/2 = 2.25 min) and 29Al (t1/2 = 6.56 min) [51,52]. Ignoring the critical 1 h buffer time will lead to a significant, unnecessary dose to any personnel handling the target.- 16.
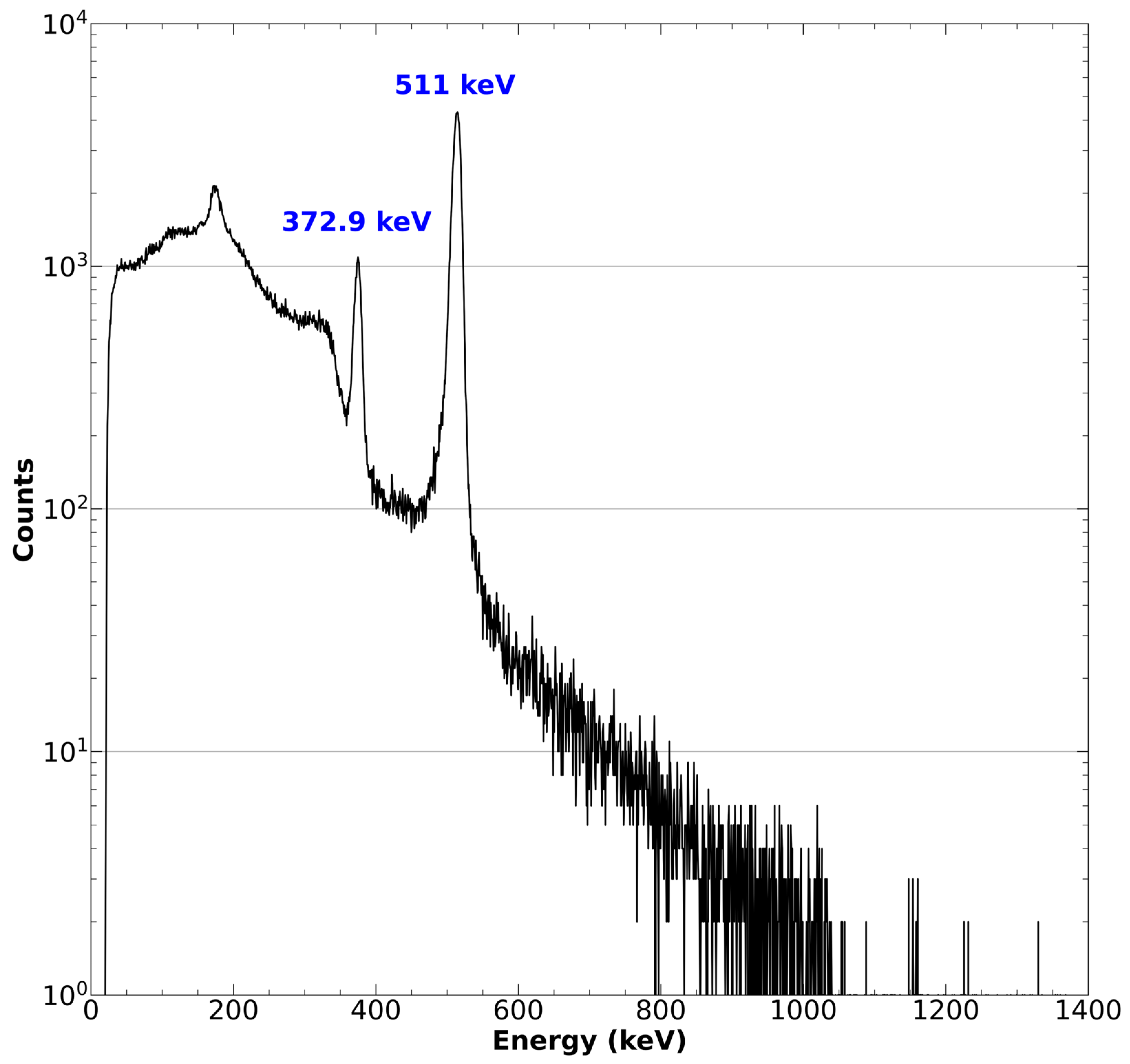
- Ensure the quantity and identity of the resultant 43Sc by measuring the activity in a well counter and confirming the identity of the radioactive source with high purity germanium (HPGe) detector or cadmium zinc telluride (CZT) gamma spectroscopy. Peaks should be observed for the characteristic 372.9 keV gamma emission as well as the 511 keV positron annihilation [41].
3.1.4. Separation of 43Sc from [42Ca]CaO—Time Needed: 1 h
CRITICAL STEP Work described in this section should be performed in a shielded fume hood or hot cell to protect staff from radioactive and chemical hazards. CRITICAL STEP Work described in this section should only be performed with metal-free or trace-analysis grade reagents. Failure to use such reagents may result in metal contamination in the 43Sc solution. Metal contaminants will compete with 43Sc for the chelation complex in the radiolabeling portion of this procedure and may result in failed conjugation with PSMA-617 [46,48]. CRITICAL STEP As this portion of the procedure involves the use of highly concentrated acids, pipette tips with filters should be utilized to minimize the risk of damage to pipettes from acid fumes. CRITICAL STEP The passing of any solution through the DGA column in Section 3.1.2. and Section 3.1.4. is done by loading the cartridge from the top and pushing the solution out from the bottom of the cartridge. Using the reverse orientation (loading from the bottom and eluting from the top) may result in the unsuccessful trapping or elution of the 43Sc product.- 17.
- Fill a conical tube with 10 mL of 0.1 M HCl and place it in a water bath heated to 95 °C. Once the 0.1 M HCl also reaches a temperature of 95 °C, it will be used in Step 24.
CRITICAL STEP While the water used in this step is not involved in any chemical reactions, it is preferable to utilize MilliQ or trace-analysis grade water to minimize metal contamination should any droplets come into contact with the HCl.- 18.
- Transfer the irradiated [42Ca]CaO/43Sc target to a 25 mL PFA beaker. Add 2 mL of 15 M HNO3 to the beaker containing the target and place the resulting solution on a hotplate preheated to 70 °C.
CRITICAL STEP Carefully monitor the temperature throughout this dissolution process. If the HNO3 begins to boil, there is a significant risk that some activity will be carried away with the vapor, resulting in a contaminated laboratory space.- 19.
- Once the [42Ca]CaO/43Sc solution is on the hot plate, add 1 mL of water every 8 min until 4 mL of water has been added, bringing the final dissolution volume to 6 mL and HNO3 concentration to 5 M. Allow the solution to remain under heat for an additional 8 min.
- 20.
- Remove the [42Ca]CaO/43Sc solution from the hotplate. Collect the solution with a micropipette, taking care to avoid any pieces of graphite foil that broke off during dissolution. Slowly load the solution into the DGA cartridge at a rate of approximately 0.2 mL/s. Collect the pass-through solution in a conical tube. As the branched DGA extractant (N,N,N’,N’-tetrakis-2-ethylhexyl-diglycolamide) has a high affinity for trivalent metals under acidic conditions, the 43Sc in the highly acidic dissolution solution should bind tightly to the DGA resin while the 42Ca target material passes through. Preserve the PFA beaker and its remaining contents in a shielded area, as they will be used in the target recovery process.
- 21.
- Assay the pass-through solution with a well counter to ensure that the radioactive 43Sc has successfully been trapped by the DGA cartridge. Place the conical tube containing the pass-through solution in a shielded area so any trace radioactivity can decay out and the solution can be retained for 42Ca recovery.
- 22.
- Gently wash the DGA column with 6 mL of 5 M HNO3. Collect and assay the washings to ensure that the 43Sc was not prematurely eluted. Again, preserve the washings in a shielded area, as they will be used in the target recovery process.
- 23.
- Gently wash the DGA column with 6 mL of 1 M HCl. Collect and assay the washings to ensure that the 43Sc was not prematurely eluted. Again, preserve the washings in a shielded area, as they will be used in the target recovery process.
- 24.
- Fill a needleless syringe with 5 mL of the 95 °C 0.1 M HCl from Step 17. Attach the DGA column to the syringe and gently elute out the [43Sc]ScCl3 product into approximately ten 500 µL aliquots, each in their own 2 mL centrifuge tube. As the introduction of a weak acid mobile phase to the DGA column greatly reduces its affinity for trivalent cations, this should result in the majority of the 43Sc atoms dissociating from the extractant, binding to the free chloride ions, and being eluted out from the column in the weak acid solution; however, a small amount of 43Sc retention in the column (<10%) is expected.
CRITICAL STEP Highly concentrated fractions of radioactivity are critical for successful radiolabeling with high molar activity, as is mandated by meaningful in vitro and in vivo experiments. As such, it is important that the elution of [43Sc]ScCl3 is performed slowly (approximately 0.1 mL/s), such that most of the activity is eluted in the first few aliquots and minimal activity is remnant in the column. CRITICAL STEP It is easy to contaminate the workspace when moving the syringe and column from one aliquot to the next. To minimize contamination, release any pressure you are placing on the plunger and move the syringe between aliquots only when it is between drops of the eluent being released from the syringe.- 25.
- Assay each aliquot in the dose calibrator to determine which have the highest concentration of radioactivity. Assay the DGA column to ensure a significant amount of activity has not remained in the column. Preserve the DGA column in a shielded area, as it will be used in the target recovery process.
- 26.
- If a large fraction of radioactivity has been retained in the DGA column (>10%), it may be necessary to elute additional fractions with 95 °C 0.1 M HCl until enough activity has been obtained for any downstream experiments. However, it is likely that these fractions will be of a lower activity concentration than those previously obtained and, therefore, suboptimal for radiolabeling.
3.1.5. Synthesis of [43Sc]Sc-PSMA-617—Time Needed: 1 h
CRITICAL STEP Work described in this section should be performed in a shielded fume hood or hot cell to protect staff from radioactive and chemical hazards. CRITICAL STEP Work described in this section should only be performed with metal-free or trace-analysis grade reagents. Failure to use such reagents may result in metal contamination in the 43Sc solution. Metal contaminants will compete with 43Sc for the chelation complex in the radiolabeling portion of this procedure and may result in failed conjugation with PSMA-617 [46,48].- 27.
- Preheat the thermal mixer to 95 °C.
- 28.
- Add an equal volume of 1 M ammonium acetate buffer (pH 5.25) to the [43Sc]ScCl3 aliquots that will be used for labeling the PSMA-617 ligand. Use a disposable pH paper to check that the pH of the buffer-activity solution is between pH 4–5, as that is the range where the conjugation of 43Sc to the DOTA chelator is most favorable. If the pH is too low, add small amounts of 0.5 M ammonium acetate buffer (pH 5.25) until the pH reaches 4.
CRITICAL STEP When using pH paper to check the pH of the buffer-activity solution, it is best to cut the paper into thin strips and then wet them with 2–5 µL using a micropipette. This ensures that contaminants are not introduced into the reaction mixture by the pH paper, and minimizes activity loss.- 29.
- Utilizing the activity data collected in Step 25, calculate the amount of PSMA-617 ligand that must be added to the buffer-activity solution to achieve the desired molar activity, according to Equation (1) below. Add the calculated amount of ligand (dissolved in water; see reagent preparation) to the buffer-activity solution.
CRITICAL STEP The maximum achievable molar activity will be heavily contingent upon the activity concentration within the reaction mixture as a consequence of Le Chatelier’s principle [53]. The amount of metal contaminants within the reaction mixture will also greatly influence the maximum achievable molar activity due to the high affinity of PSMA-617’s DOTA chelation complex for heavy metals such as iron, copper, and zinc [54]. As such, one should consider these two factors when selecting a target molar activity, and, even more so, as potential explanations if a radiolabeling reaction fails at a given target molar activity. Consequently, we recommend users of this protocol begin their implementation of this protocol with a low target molar activity (for example, 1 MBq/nmol), and then incrementally increase the target molar activity with each sequential experiment.- 30.
- Place the reaction mixture into the preheated thermal mixer. Leave the temperature at 95 °C and set the mixing frequency to ~500 RPM. Allow the reaction to occur under these conditions for 40 min.
- 31.
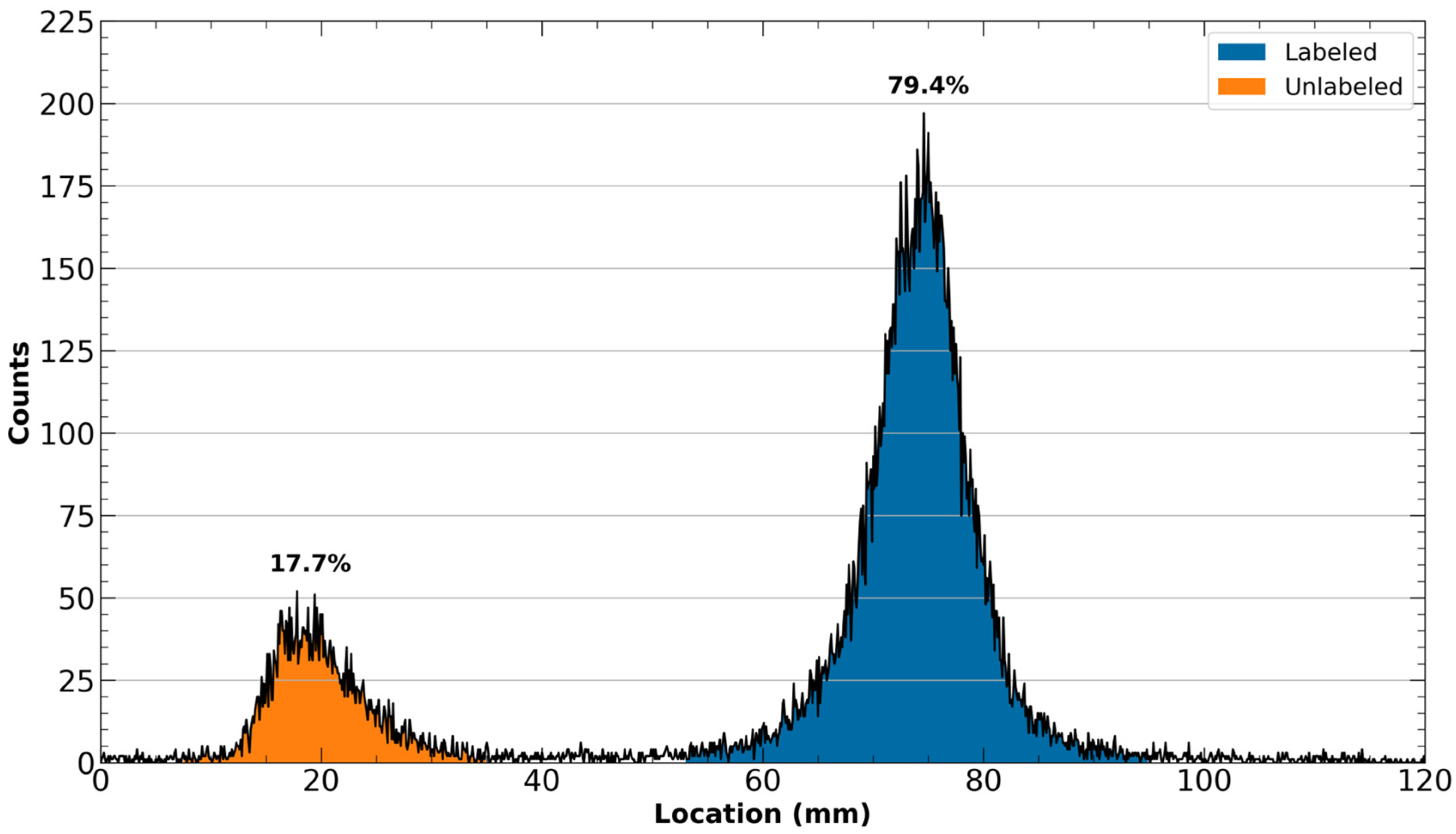
- Once the reaction time has elapsed, check that [43Sc]Sc-PSMA-617 has been formed by spotting on an iTLC-SG paper with approximately 2 µL of the reaction mixture. Develop the spotted iTLC paper in a mobile phase consisting of a 1:1 ratio of methanol and 1 M ammonium formate. Once developed, analyze the iTLC plate with a radioTLC scanner. Unlabeled 43Sc should remain near the spotting line, whereas [43Sc]Sc-PSMA-617 will migrate with the solvent front.
3.1.6. Purification and Quality Control of [43Sc]Sc-PSMA-617—Time Needed: 20 min
CRITICAL STEP Work described in this section should be performed in a shielded fume hood or hot cell to protect staff from radioactive and chemical hazards.- 32.
- Condition a Waters Oasis HLB light cartridge (30 mg sorbent) by slowly pushing 3 mL of ethanol followed by 6 mL of water at a rate of approximately 1.0 mL/s.
- 33.
- Load the radiolabeled solution into the conditioned Oasis cartridge and slowly pass it through. Collect and assay the pass-through in a well counter to ensure that the [43Sc]Sc-PSMA-617 has been retained on the cartridge. Any unlabeled 43Sc should be eluted in the pass-through solution and subsequent wash described in Step 33.
- 34.
- Wash the Oasis cartridge with 2 mL of water. Collect and assay the washings to ensure that the [43Sc]Sc-PSMA-617 has not prematurely eluted.
- 35.
- Slowly elute the [43Sc]Sc-PSMA-617 solution with 300 µL of ethanol at an approximate rate of 0.05 mL/s. If the activity will be utilized for an animal injection, it is recommended to elute into a screw top septum vial (e.g., 4 mL) as is necessary for steps 37–41.
- 36.
- Repeat Step 30 with the now-purified [43Sc]Sc-PSMA-617 solution to ensure that all unlabeled 43Sc has been removed. The subsequent iTLC should now have only a single peak that moves with the solvent front.
- 37.
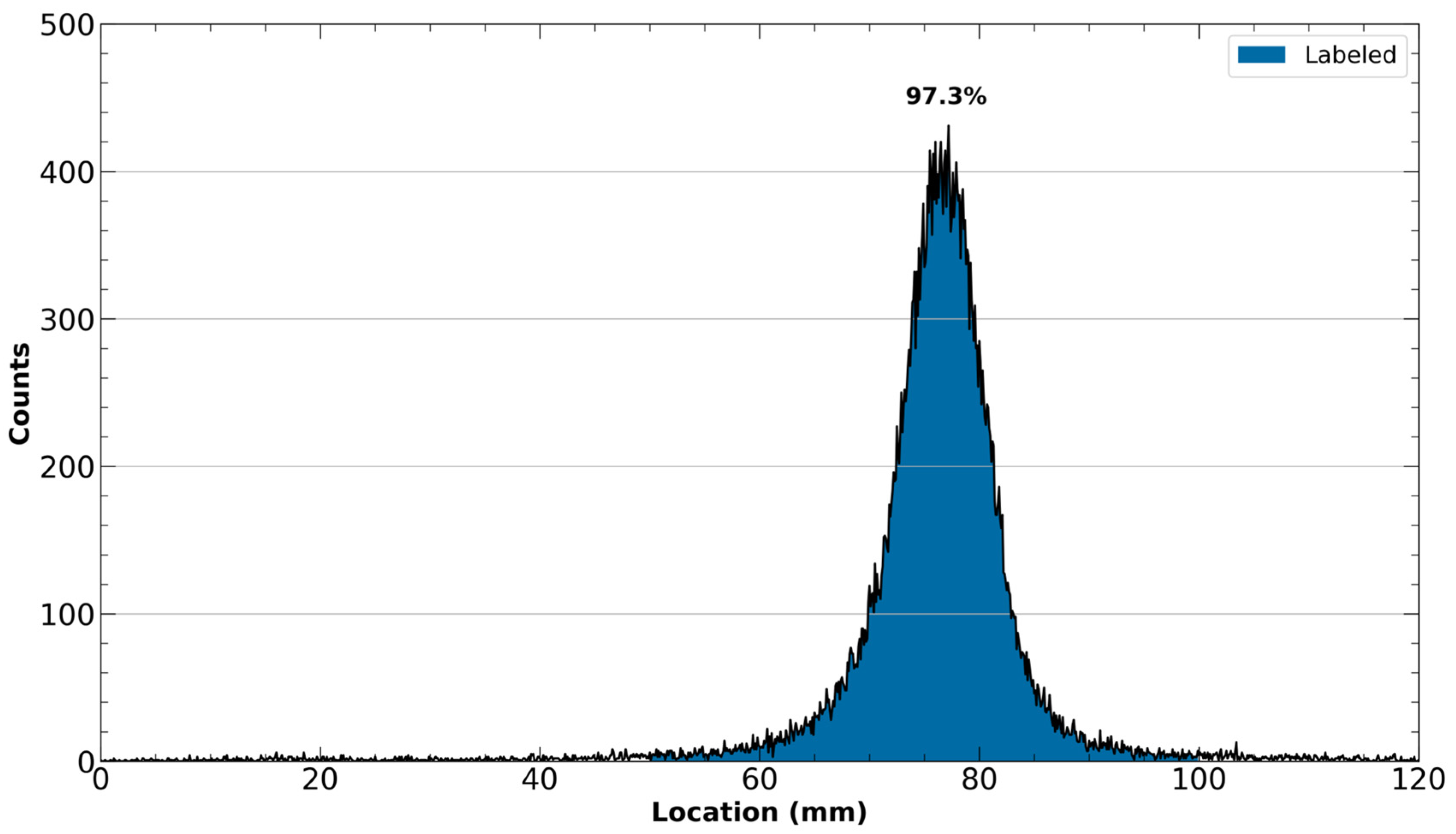
- If reformulation of the [43Sc]Sc-PSMA-617 is unnecessary, such as in cellular studies where the ethanol will be greatly diluted, transfer 25 µL of the final purified solution into a small V-vial. From this V-vial, inject 5 µL into the HPLC system for radio-HPLC analysis. The resultant chromatogram can be compared to a standard curve generated by the UV trace of a reference solution of cold PSMA-617 to calculate the true molar activity of the final solution.
3.1.7. Reformulation of [43Sc]Sc-PSMA-617—Time Needed: 20 min
CRITICAL STEP Work described in this section should be performed in a shielded fume hood or hot cell to protect staff from radioactive and chemical hazards.- 38.
- Preheat a hot plate to 60 °C.
CRITICAL STEP It is critical that the hotplate is preheated to the target temperature prior to placing the ethanol solution on it. If the hotplate is instead heated while the target solution is in contact with it, temperature overshoot can occur, causing the solution to boil too rapidly, thus yielding a buildup of excessive pressure within the vial and increasing the surface area the radioactive solution comes into contact with (reducing downstream yield).- 39.
- Transfer the ethanol solution containing [43Sc]Sc-PSMA-617 produced in Step 34 to a screw top septum vial if it is not already in one. Place the vial on the preheated hotplate. Attach a 20-gauge needle to a nitrogen line and carefully insert it into the septum, ensuring that the needle is not touching the radioactive solution. Insert another 18-gauge needle into the septum for venting. Turn on a gentle flow of nitrogen and evaporate the ethanol medium to near dryness.
- 40.
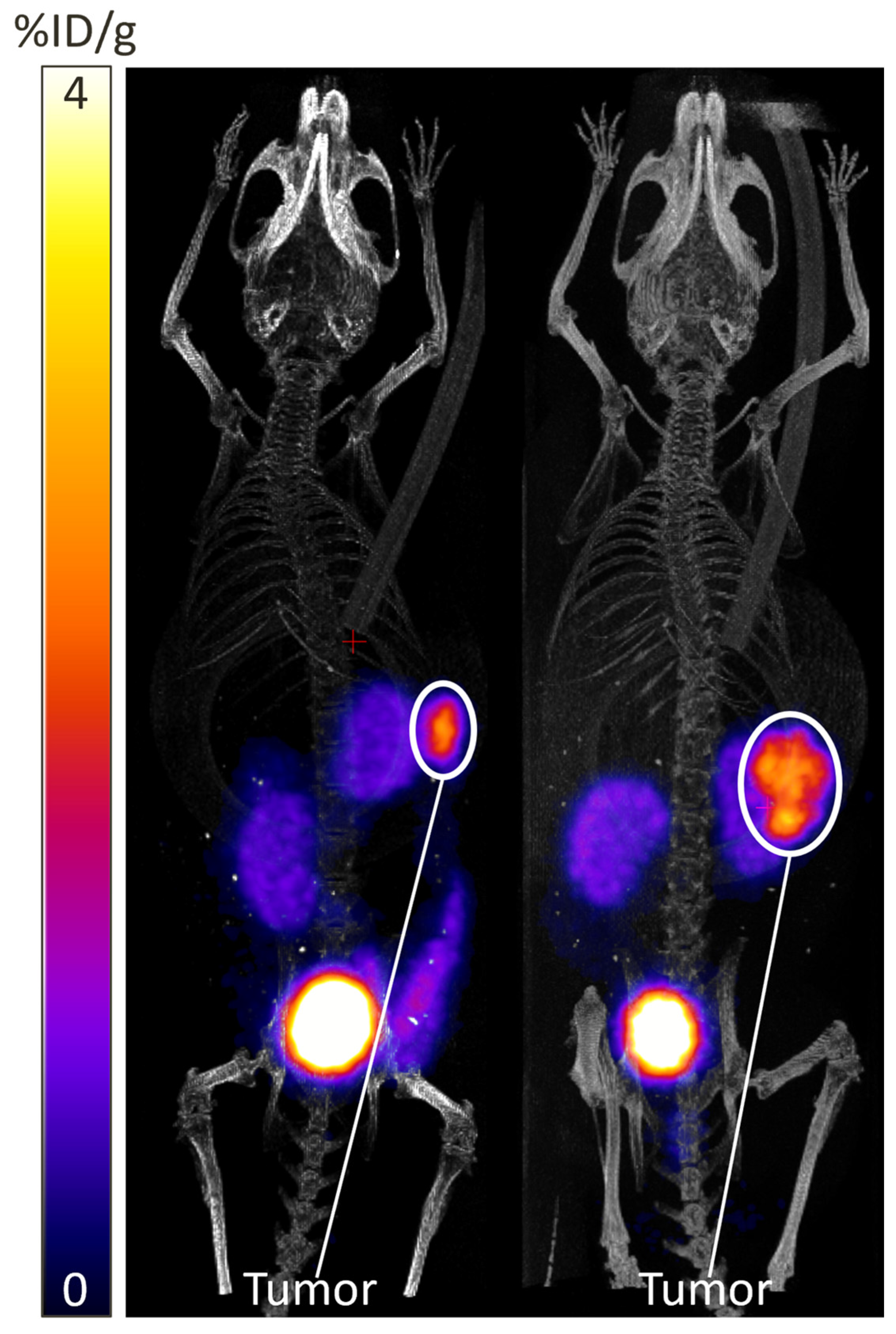
- Use a needle and syringe to add the desired reconstituting medium to the vial at the desired activity concentration. For PET imaging, we recommend using a ratio of approximately 0.5 µL of PBS for every 0.037 MBq of radioactivity.
- 41.
- Utilize a disposable pH paper to check that the pH of the final dose is near the blood pH of the experimental group (pH ~7–7.4 in mice). If the measured pH falls outside the physiological range, continue to add PBS until the pH becomes a safe level for in vivo use.
CRITICAL STEP As the final reformulated dose is intended for in vivo use, it is critical that the pH is checked before in vivo application after every synthesis. Injection of a radiopharmaceutical in a non-physiological solvent could cause suffering, physical harm, and death in the experimental subject [55,56]. This is especially true when conducting in vivo studies with animals that have a low blood volume, such as mice, or animals with impaired renal function, as is the case in animals with tumor xenografts [56]. CRITICAL STEP When using pH paper to check the pH of the buffer-activity solution, it is best to cut the paper into thin strips and then wet them with 2–5 µL using a micropipette. This ensures that contaminants are not introduced into the reaction mixture by the pH paper, and minimizes activity loss.- 42.
- Analyze an aliquot of the reconstituted solution by radio-HPLC as described in Step 36.
3.2. Recycling of 42Ca Target Materials—Time Needed: 12 h
CRITICAL STEP Prior to beginning the recovery procedure, assay the recovery materials collected in Steps 20–25 and ensure that they are no longer radioactive and thus safe for handling. Performing target recovery with radioactive materials will lead to an unnecessary dose to staff and potential contamination of the laboratory space. CRITICAL STEP It is recommended to combine and recover the 42Ca from multiple productions simultaneously rather than performing a recovery after each individual production. This is because each transfer of the 42Ca recovery materials that is performed will result in the loss of some 42Ca. By minimizing the number of times the procedure is performed, the number of transfers is minimized as well.- Wash the used DGA column from Step 25 in Section 3.1.4. with 10 mL of 3 M HCl. Collect the washings in an approximately 200 mL beaker.
- Combine the DGA pass-through, 1 M HCl wash, and 5 M HNO3 wash from Steps 21–23 of Section 3.1.4. in the 200 mL beaker. Also wash each of the conical tubes that contained these solutions with 2 mL of metal-free water and collect those washings in the 200 mL beaker.
- Wash the PFA beaker that was used in Step 20 of Section 3.1.4. with 2 mL of metal-free water and collect the washings in the 200 mL beaker.
- OPTIONAL STEP Store any used recovery materials from the previous four steps such that they are available if a secondary recovery is necessary.
- Add a large stir bar to the 200 mL beaker and place it on a hot plate set to approximately 150 °C with gentle stirring. Carefully evaporate the solution to dryness.
CRITICAL STEP It is important to evaporate the solution slowly, as vigorous boiling can cause portions of the recovery solution to splash outside of the beaker, resulting in a loss of 42Ca material. CRITICAL STEP It is critical to quickly remove the dried solution from the hot plate as soon as it reaches complete dryness. Should the dry solution be left on the hot plate too long, it can undergo incomplete combustion, yielding a charred substance. This charring will inhibit the downstream recovery processes.- 6.
- Redissolve the dried solution in 5 mL of 3 M HCl and repeat the previous step (Step 5) of this procedure.
- 7.
- Once fully dried, add 20 mL of water to the 200 mL beaker to redissolve the calcium.
- 8.
- Adjust the pH of the calcium solution to pH 4.5–5 using 2.5% ammonium hydroxide and 1 M HCl.
- 9.
- Add 20 mL of 0.3 M ammonium oxalate to the pH adjusted solution, yielding calcium oxalate. Upon adding the ammonium oxalate, the solution should turn an opaque, cloudy white color.
- 10.
- Allow the calcium oxalate solution to stand until all particulates have settled to the bottom of the 200 mL beaker.
- 11.
- Filter the calcium oxalate solution through a Buchner funnel and two stacked 8 µm pore-size ashless Wattman 42 filter papers under vacuum. The filtrate should be collected in a round bottom flask and re-run through the filter paper until the filtrate is completely clear.
- 12.
- Allow the vacuum to continue to pull on the filter paper until dried. Once fully dried, remove the filter papers and place them in a crucible for calcination.
- 13.
- Proceed with the calcination process described in Steps 2–4 of Section 3.1.1.
4. Expected Results
4.1. Synthesis of Injectable [43Sc]Sc-PSMA-617 from [42Ca]CaCO3
4.1.1. Cyclotron Production of 43Sc
4.1.2. Separation of 43Sc from [42Ca]CaO
4.1.3. Synthesis of [43Sc]Sc-PSMA-617
4.1.4. Purification and Quality Control of [43Sc]Sc-PSMA-617
4.1.5. Reformulation of [43Sc]Sc-PSMA-617
4.2. Recycling of 42Ca Target Materials
5. Reagents Setup
5.1. Production of [43Sc]Sc-PSMA-617 Using Cyclotron Produced 43Sc
- 5 M HNO3 (metal free)
| Reagent | Final Concentration | Amount |
| Metal-Free HNO3 (15 M) | 5 M | 5 mL |
| Trace Analysis Water | 10 mL | |
| Total | 15 mL |
- 2.
- 2.5 M HNO3 (metal free)
| Reagent | Final Concentration | Amount |
| Metal-Free HNO3 (5 M) | 2.5 M | 3 mL |
| Trace Analysis Water | 3 mL | |
| Total | 6 mL |
- 3.
- 1 M HCl (metal free)
| Reagent | Final Concentration | Amount |
| Metal-Free HCl (12 M) | 1 M | 1 mL |
| Trace Analysis Water | 11 mL | |
| Total | 12 mL |
- 4.
- 0.1 M HCl (metal free)
| Reagent | Final Concentration | Amount |
| Metal-Free HCl (1 M) | 0.1 M | 1 mL |
| Trace Analysis Water | 9 mL | |
| Total | 10 mL |
- 5.
- 1 M Ammonium Acetate Buffer (pH 5.25)
| Reagent | Final Concentration | Amount |
| Ammonium Acetate | ~1 M | 0.78 g |
| Trace Analysis Water | 10 mL | |
| Acetic Acid | Variable 1 | Variable 1 |
| Total | >10 mL | |
| 1 Acetic acid is added to the aqueous ammonium acetate solution until the solution has reached pH 5.25. Consequently, the amount added and the concentration of acetic acid in the final reagent are not fixed quantities. As such, it is recommended that acetic acid be gradually added to the aqueous ammonium acetate in 100 μL increments and that the pH is checked with a benchtop pH meter/electrode after each increment until the desired pH is reached. | ||
- 6.
- PSMA-617 (aqueous)
| Reagent | Final Concentration | Amount |
| PSMA-617 | 0.96 mM | 50 μg |
| Trace Analysis Water | 50 μL | |
| Total | 50 μL |
5.2. Recycling of 42Ca Target Materials
- 3 M HCl (metal free)
| Reagent | Final Concentration | Amount |
| Metal-Free HCl (12 M) | 3 M | 6 mL |
| Trace Analysis Water | 18 mL | |
| Total | 24 mL |
- 2.
- 1 M HCl (metal free)
| Reagent | Final Concentration | Amount |
| Metal-Free HCl (3 M) | 1 M | 4 mL |
| Trace Analysis Water | 8 mL | |
| Total | 12 mL |
- 3.
- 0.3 M Ammonium Oxalate
| Reagent | Final Concentration | Amount |
| Ammonium Oxalate | 0.3 M | 0.45 g |
| Trace Analysis Water | 20 mL | |
| Total | 20 mL |
- 4.
- 0.64 M Ammonium Hydroxide
| Reagent | Final Concentration | Amount |
| Ammonium Hydroxide (5.65 M) | 0.64 M | 1.36 mL |
| Trace Analysis Water | 11.64 mL | |
| Total | 12 mL |
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gomes Marin, J.F.; Nunes, R.F.; Coutinho, A.M.; Zaniboni, E.C.; Costa, L.B.; Barbosa, F.G.; Queiroz, M.A.; Cerri, G.G.; Buchpiguel, C.A. Theranostics in Nuclear Medicine: Emerging and Re-Emerging Integrated Imaging and Therapies in the Era of Precision Oncology. Radiographics 2020, 40, 1715–1740. [Google Scholar] [CrossRef] [PubMed]
- Langbein, T.; Weber, W.A.; Eiber, M. Future of Theranostics: An Outlook on Precision Oncology in Nuclear Medicine. J. Nucl. Med. 2019, 60, 13S–19S. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, E.; Bodei, L.; Morris, M.J. The Role of Theranostics in Prostate Cancer. Semin. Radiat. Oncol. 2021, 31, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, A.; Eppard, E.; Kürpig, S.; Bundschuh, R.; Schönberger, S.; Gonzalez-Carmona, M.; Feldmann, G.; Ahmadzadehfar, H.; Essler, M. Theranostics in Nuclear Medicine Practice. OncoTargets Ther. 2017, 10, 4821–4828. [Google Scholar] [CrossRef]
- Kuo, P.H.; Morris, M.J.; Hesterman, J.; Kendi, A.T.; Rahbar, K.; Wei, X.X.; Fang, B.; Adra, N.; Garje, R.; Michalski, J.M.; et al. Quantitative68 Ga-PSMA-11 PET and Clinical Outcomes in Metastatic Castration-Resistant Prostate Cancer Following177 Lu-PSMA-617 (VISION Trial). Radiology 2024, 312, e233460. [Google Scholar] [CrossRef]
- Kuo, P.; Hesterman, J.; Rahbar, K.; Kendi, A.T.; Wei, X.X.; Fang, B.; Adra, N.; Armstrong, A.J.; Garje, R.; Michalski, J.M.; et al. [68Ga]Ga-PSMA-11 PET Baseline Imaging as a Prognostic Tool for Clinical Outcomes to [177Lu]Lu-PSMA-617 in Patients with mCRPC: A VISION Substudy. J. Clin. Oncol. 2022, 40, 5002. [Google Scholar] [CrossRef]
- Groener, D.; Schneider, S.; Baumgarten, J.; Happel, C.; Klimek, K.; Mader, N.; Nguyen Ngoc, C.; Wichert, J.; Mandel, P.; Tselis, N.; et al. Baseline [68Ga]Ga-PSMA-11 PET/CT before [177Lu]Lu-PSMA-617 Radioligand Therapy: Value of PSMA-Uptake Thresholds in Predicting Targetable Lesions. Cancers 2023, 15, 473. [Google Scholar] [CrossRef]
- Sartor, O.; De Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Peters, S.M.B.; Hofferber, R.; Privé, B.M.; de Bakker, M.; Gotthardt, M.; Janssen, M.; de Lange, F.; Muselaers, C.H.J.; Mehra, N.; Witjes, J.A.; et al. [68Ga]Ga-PSMA-11 PET Imaging as a Predictor for Absorbed Doses in Organs at Risk and Small Lesions in [177Lu]Lu-PSMA-617 Treatment. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1101–1112. [Google Scholar] [CrossRef]
- Kabasakal, L.; AbuQbeitah, M.; Aygün, A.; Yeyin, N.; Ocak, M.; Demirci, E.; Toklu, T. Pre-Therapeutic Dosimetry of Normal Organs and Tissues of 177Lu-PSMA-617 Prostate-Specific Membrane Antigen (PSMA) Inhibitor in Patients with Castration-Resistant Prostate Cancer. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1976–1983. [Google Scholar] [CrossRef]
- Silberstein, E.B. Radioiodine: The Classic Theranostic Agent. Semin. Nucl. Med. 2012, 42, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Ehlerding, E.B.; Ferreira, C.A.; Aluicio-Sarduy, E.; Jiang, D.; Lee, H.J.; Theuer, C.P.; Engle, J.W.; Cai, W. 86/90Y-Based Theranostics Targeting Angiogenesis in a Murine Breast Cancer Model. Mol. Pharm. 2018, 15, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Krasnovskaya, O.O.; Abramchuck, D.; Erofeev, A.; Gorelkin, P.; Kuznetsov, A.; Shemukhin, A.; Beloglazkina, E.K. Recent Advances in 64Cu/67Cu-Based Radiopharmaceuticals. Int. J. Mol. Sci. 2023, 24, 9154. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sagastume, E.A.; Lee, D.; McAlister, D.; DeGraffenreid, A.J.; Olewine, K.R.; Graves, S.; Copping, R.; Mirzadeh, S.; Zimmerman, B.E.; et al. 203/212Pb Theranostic Radiopharmaceuticals for Image-Guided Radionuclide Therapy for Cancer. Curr. Med. Chem. 2020, 27, 7003–7031. [Google Scholar] [CrossRef]
- Müller, C.; Domnanich, K.A.; Umbricht, C.A.; van der Meulen, N.P. Scandium and Terbium Radionuclides for Radiotheranostics: Current State of Development towards Clinical Application. Br. J. Radiol. 2018, 91, 20180074. [Google Scholar] [CrossRef]
- Naskar, N.; Lahiri, S. Theranostic Terbium Radioisotopes: Challenges in Production for Clinical Application. Front. Med. 2021, 8, 675014. [Google Scholar] [CrossRef]
- Currie, G.M.; Rohren, E.M. Sharpening the Blade of Precision Theranostics. Semin. Nucl. Med. 2025, in press. [Google Scholar] [CrossRef]
- Huclier-Markai, S.; Alliot, C.; Kerdjoudj, R.; Mougin-Degraef, M.; Chouin, N.; Haddad, F. Promising Scandium Radionuclides for Nuclear Medicine: A Review on the Production and Chemistry up to In Vivo Proofs of Concept. Cancer Biother. Radiopharm. 2018, 33, 316–329. [Google Scholar] [CrossRef]
- Kilian, K.; Pyrzyńska, K. Scandium Radioisotopes—Toward New Targets and Imaging Modalities. Molecules 2023, 28, 7668. [Google Scholar] [CrossRef]
- Muller, C.; Bunka, M.; Haller, S.; Koster, U.; Groehn, V.; Bernhardt, P.; van der Meulen, N.; Turler, A.; Schibli, R. Promising Prospects for 44Sc-/47Sc-Based Theragnostics: Application of 47Sc for Radionuclide Tumor Therapy in Mice. J. Nucl. Med. 2014, 55, 1658–1664. [Google Scholar] [CrossRef]
- Singh, B.; Chen, J. Nuclear Data Sheets for A = 43. Nucl. Data Sheets 2015, 126, 1–150. [Google Scholar] [CrossRef]
- Chen, J.; Singh, B. Nuclear Structure and Decay Data for A = 44 Isobars. Nucl. Data Sheets 2023, 190, 1–318. [Google Scholar] [CrossRef]
- Martin, C.C.; Christian, B.T.; Satter, M.R.; Nickerson, L.D.H.; Nickles, R.J. Quantitative PET with Positron Emitters That Emit Prompt Gamma Rays. IEEE Trans. Med. Imaging 1995, 14, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.V.; Mendes, B.M.; Paixão, L.; Gnesin, S.; Müller, C.; van der Meulen, N.P.; Strobel, K.; Fonseca, T.C.F.; Lima, T.V.M. Comparison of the Dosimetry of Scandium-43 and Scandium-44 Patient Organ Doses in Relation to Commonly Used Gallium-68 for Imaging Neuroendocrine Tumours. EJNMMI Phys. 2024, 11, 61. [Google Scholar] [CrossRef]
- Burrows, T.W. Nuclear Data Sheets for A = 47. Nucl. Data Sheets 2007, 108, 923–1056. [Google Scholar] [CrossRef]
- Mikolajczak, R.; Huclier-Markai, S.; Alliot, C.; Haddad, F.; Szikra, D.; Forgacs, V.; Garnuszek, P. Production of Scandium Radionuclides for Theranostic Applications: Towards Standardization of Quality Requirements. EJNMMI Radiopharm. Chem. 2021, 6, 19. [Google Scholar] [CrossRef]
- Cingoranelli, S.J.; Bartels, J.L.; Kankanamalage, P.H.A.; Loveless, C.S.; Rotsch, D.A.; Lapi, S.E. Production and Purification of 43Sc and 47Sc from Enriched [46Ti]TiO2 and [50Ti]TiO2 Targets. Sci. Rep. 2023, 13, 22683. [Google Scholar] [CrossRef]
- Chernysheva, M.; Loveless, S.C.; Brossard, T.; Becker, K.; Cingoranelli, S.; Aluicio-Sarduy, E.; Song, J.; Ellison, P.; Nolen, J.; Rotsch, D.A.; et al. Accelerator Production of Scandium Radioisotopes: Sc-43, Sc-44, and Sc-47. Curr. Radiopharm. 2021, 14, 359–373. [Google Scholar] [CrossRef]
- Becker, K.V.; Aluicio-Sarduy, E.; Bradshaw, T.; Hurley, S.A.; Olson, A.P.; Barrett, K.E.; Batterton, J.; Ellison, P.A.; Barnhart, T.E.; Pirasteh, A.; et al. Cyclotron Production of 43Sc and 44gSc from Enriched 42CaO, 43CaO, and 44CaO Targets. Front. Chem. 2023, 11, 1167783. [Google Scholar] [CrossRef]
- Minegishi, K.; Nagatsu, K.; Fukada, M.; Suzuki, H.; Ohya, T.; Zhang, M.-R. Production of Scandium-43 and -47 from a Powdery Calcium Oxide Target via the Nat/44Ca(α,x)-Channel. Appl. Radiat. Isot. 2016, 116, 8–12. [Google Scholar] [CrossRef]
- Rotsch, D.A.; Brown, M.A.; Nolen, J.A.; Brossard, T.; Henning, W.F.; Chemerisov, S.D.; Gromov, R.G.; Greene, J. Electron Linear Accelerator Production and Purification of Scandium-47 from Titanium Dioxide Targets. Appl. Radiat. Isot. 2018, 131, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Loveless, C.S.; Blanco, J.R.; Diehl, G.L.; Elbahrawi, R.T.; Carzaniga, T.S.; Braccini, S.; Lapi, S.E. Cyclotron Production and Separation of Scandium Radionuclides from Natural Titanium Metal and Titanium Dioxide Targets. J. Nucl. Med. 2021, 62, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Loveless, C.S.; Radford, L.L.; Ferran, S.J.; Queern, S.L.; Shepherd, M.R.; Lapi, S.E. Photonuclear Production, Chemistry, and in Vitro Evaluation of the Theranostic Radionuclide 47Sc. EJNMMI Res 2019, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Misiak, R.; Walczak, R.; Wąs, B.; Bartyzel, M.; Mietelski, J.W.; Bilewicz, A. 47Sc Production Development by Cyclotron Irradiation of 48Ca. J. Radioanal. Nucl. Chem. 2017, 313, 429–434. [Google Scholar] [CrossRef]
- Abel, E.P.; Domnanich, K.; Clause, H.K.; Kalman, C.; Walker, W.; Shusterman, J.A.; Greene, J.; Gott, M.; Severin, G.W. Production, Collection, and Purification of 47Ca for the Generation of 47Sc through Isotope Harvesting at the National Superconducting Cyclotron Laboratory. ACS Omega 2020, 5, 27864–27872. [Google Scholar] [CrossRef]
- Van Der Meulen, N.P.; Bunka, M.; Domnanich, K.A.; Müller, C.; Haller, S.; Vermeulen, C.; Türler, A.; Schibli, R. Cyclotron Production of 44Sc: From Bench to Bedside. Nucl. Med. Biol. 2015, 42, 745–751. [Google Scholar] [CrossRef]
- Domnanich, K.A.; Eichler, R.; Muller, C.; Jordi, S.; Yakusheva, V.; Braccini, S.; Behe, M.; Schibli, R.; Turler, A.; van der Meulen, N.P. Production and Separation of 43Sc for Radiopharmaceutical Purposes. EJNMMI Radiopharm. Chem. 2017, 2, 14. [Google Scholar] [CrossRef]
- Notni, J.; Wester, H. Re-thinking the Role of Radiometal Isotopes: Towards a Future Concept for Theranostic Radiopharmaceuticals. J. Label. Compd. Radiopharm. 2018, 61, 141–153. [Google Scholar] [CrossRef]
- Tuerler, A.; Van Der Meulen, N.; Bunka, M. Production of 43Sc Radionuclide and Radiopharmaceuticals Thereof for Use in Positron Emission Tomography. U.S. Patent No. 20170087260, 30 March 2017. [Google Scholar]
- Duchemin, C.; Guertin, A.; Haddad, F.; Michel, N.; Métivier, V. Production of Scandium-44m and Scandium-44g with Deuterons on Calcium-44: Cross Section Measurements and Production Yield Calculations. Phys. Med. Biol. 2015, 60, 6847–6864. [Google Scholar] [CrossRef]
- Chadwick, M.B.; Obložinský, P.; Herman, M.; Greene, N.M.; McKnight, R.D.; Smith, D.L.; Young, P.G.; MacFarlane, R.E.; Hale, G.M.; Frankle, S.C.; et al. ENDF/B-VII.0: Next Generation Evaluated Nuclear Data Library for Nuclear Science and Technology. Nucl. Data Sheets 2006, 107, 2931–3060. [Google Scholar] [CrossRef]
- Koning, A.J.; Rochman, D.; Sublet, J.C.; Dzysiuk, N.; Fleming, M.; van der Marck, S. TENDL: Complete Nuclear Data Library for Innovative Nuclear Science and Technology. Nucl. Data Sheets 2019, 155, 1–55. [Google Scholar] [CrossRef]
- Huclier-Markai, S.; Kerdjoudj, R.; Alliot, C.; Bonraisin, A.C.; Michel, N.; Haddad, F.; Barbet, J. Optimization of Reaction Conditions for the Radiolabeling of DOTA and DOTA-Peptide with 44m/44Sc and Experimental Evidence of the Feasibility of an in Vivo PET Generator. Nucl. Med. Biol. 2014, 41, e36–e43. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, I.; Lefkaritis, G.; Georgiades, S.N.; Pashalidis, I.; Kontoghiorghes, G.J. Towards Clinical Development of Scandium Radioisotope Complexes for Use in Nuclear Medicine: Encouraging Prospects with the Chelator 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid (DOTA) and Its Analogues. Int. J. Mol. Sci. 2024, 25, 5954. [Google Scholar] [CrossRef] [PubMed]
- Kerdjoudj, R.; Pniok, M.; Alliot, C.; Kubíček, V.; Havlíčková, J.; Rösch, F.; Hermann, P.; Huclier-Markai, S. Scandium(iii) Complexes of Monophosphorus Acid DOTA Analogues: A Thermodynamic and Radiolabelling Study with 44Sc from Cyclotron and from a 44Ti/44Sc Generator. Dalton Trans. 2016, 45, 1398–1409. [Google Scholar] [CrossRef]
- Kovács, A. Metal–Ligand Interactions in Scandium Complexes with Radiopharmaceutical Applications. Inorg. Chem. 2023, 62, 20733–20744. [Google Scholar] [CrossRef]
- Pniok, M.; Kubíček, V.; Havlíčková, J.; Kotek, J.; Sabatie-Gogová, A.; Plutnar, J.; Huclier-Markai, S.; Hermann, P. Thermodynamic and Kinetic Study of Scandium(III) Complexes of DTPA and DOTA: A Step Toward Scandium Radiopharmaceuticals. Chem. Eur. J. 2014, 20, 7944–7955. [Google Scholar] [CrossRef]
- Walczak, R.; Gawęda, W.; Dudek, J.; Choiński, J.; Bilewicz, A. Influence of Metal Ions on the 44Sc-Labeling of DOTATATE. J. Radioanal. Nucl. Chem. 2019, 322, 249–254. [Google Scholar] [CrossRef]
- Majkowska-Pilip, A.; Bilewicz, A. Macrocyclic Complexes of Scandium Radionuclides as Precursors for Diagnostic and Therapeutic Radiopharmaceuticals. J. Inorg. Biochem. 2011, 105, 313–320. [Google Scholar] [CrossRef]
- Meier, J.P.; Zhang, H.J.; Freifelder, R.; Bhuiyan, M.; Selman, P.; Mendez, M.; Kankanamalage, P.H.A.; Brossard, T.; Pusateri, A.; Tsai, H.-M.; et al. Accelerator-Based Production of Scandium Radioisotopes for Applications in Prostate Cancer: Toward Building a Pipeline for Rapid Development of Novel Theranostics. Molecules 2023, 28, 6041. [Google Scholar] [CrossRef]
- Fang, P.-W.; Wang, K.-W.; Hsieh, Y.-I.; Huang, J.-C.; Sheu, R.-J. Characteristics of Neutron Production and Concrete Activation in Cyclotron Vaults for Self-Shielded and Non-Self-Shielded Facilities. Radiat. Phys. Chem. 2022, 201, 110448. [Google Scholar] [CrossRef]
- Mukherjee, B.; Khachan, J. Component Activation of a High Current Radioisotope Production Medical Cyclotron. In Proceedings of the 18th International Conference on Cyclotrons and Their Applications, Giardini Naxos, Italy, 30 September–5 October 2007. [Google Scholar]
- Holland, J.P. Chemical Kinetics of Radiolabelling Reactions. Chem. Eur. J. 2018, 24, 16472–16483. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Breeman, W.A.P.; Klette, I.; Gottschaldt, M.; Odparlik, A.; Baehre, M.; Tworowska, I.; Schultz, M.K. Radiolabeling of DOTA-like Conjugated Peptides with Generator-Produced 68Ga and Using NaCl-Based Cationic Elution Method. Nat. Protoc. 2016, 11, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Iversen, N.K.; Malte, H.; Baatrup, E.; Wang, T. The Normal Acid–Base Status of Mice. Respir. Physiol. Neurobiol. 2012, 180, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of Substances to Laboratory Animals: Routes of Administration and Factors to Consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [Google Scholar]
- Julian, W.; Sergeeva, O.; Cao, W.; Wu, C.; Erokwu, B.; Flask, C.; Zhang, L.; Wang, X.; Basilion, J.; Yang, S.; et al. Searching for Protein Off-Targets of Prostate-Specific Membrane Antigen-Targeting Radioligands in the Salivary Glands. Cancer Biother. Radiopharm. 2024, 39, 721–732. [Google Scholar] [CrossRef]
- Ghosh, A.; Wang, X.; Klein, E.; Heston, W.D.W. Novel Role of Prostate-Specific Membrane Antigen in Suppressing Prostate Cancer Invasiveness. Cancer Res. 2005, 65, 727–731. [Google Scholar] [CrossRef]
- Ruigrok, E.A.M.; Van Vliet, N.; Dalm, S.U.; De Blois, E.; Van Gent, D.C.; Haeck, J.; De Ridder, C.; Stuurman, D.; Konijnenberg, M.W.; Van Weerden, W.M.; et al. Extensive Preclinical Evaluation of Lutetium-177-Labeled PSMA-Specific Tracers for Prostate Cancer Radionuclide Therapy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1339–1350. [Google Scholar] [CrossRef]
- Gorges, T.M.; Riethdorf, S.; von Ahsen, O.; Nastały, P.; Röck, K.; Boede, M.; Peine, S.; Kuske, A.; Schmid, E.; Kneip, C.; et al. Heterogeneous PSMA Expression on Circulating Tumor Cells: A Potential Basis for Stratification and Monitoring of PSMA-Directed Therapies in Prostate Cancer. Oncotarget 2016, 7, 34930–34941. [Google Scholar] [CrossRef]
- Sheehan, B.; Neeb, A.; Buroni, L.; Paschalis, A.; Riisnaes, R.; Gurel, B.; Gil, V.; Miranda, S.; Crespo, M.; Guo, C.; et al. Prostate-Specific Membrane Antigen Expression and Response to DNA Damaging Agents in Prostate Cancer. Clin. Cancer Res. 2022, 28, 3104–3115. [Google Scholar] [CrossRef]
- Herrmann, K.; Rahbar, K.; Eiber, M.; Sparks, R.; Baca, N.; Krause, B.J.; Lassmann, M.; Jentzen, W.; Tang, J.; Chicco, D.; et al. Renal and Multiorgan Safety of 177Lu-PSMA-617 in Patients with Metastatic Castration-Resistant Prostate Cancer in the VISION Dosimetry Substudy. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2024, 65, 71–78. [Google Scholar] [CrossRef]
- Schuchardt, C.; Zhang, J.; Kulkarni, H.R.; Chen, X.; Müller, D.; Baum, R.P. Prostate-Specific Membrane Antigen Radioligand Therapy Using 177Lu-PSMA I&T and 177Lu-PSMA-617 in Patients with Metastatic Castration-Resistant Prostate Cancer: Comparison of Safety, Biodistribution, and Dosimetry. J. Nucl. Med. 2022, 63, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Hofman, M.; McIntosh, L.; Buteau, J.P.; Ravi Kumar, A. Radiation Dosimetry in 177Lu-PSMA-617 Therapy. Semin. Nucl. Med. 2022, 52, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Piron, S.; Verhoeven, J.; De Coster, E.; Descamps, B.; Kersemans, K.; Pieters, L.; Vral, A.; Vanhove, C.; De Vos, F. Impact of the Molar Activity and PSMA Expression Level on [18F]AlF-PSMA-11 Uptake in Prostate Cancer. Sci. Rep. 2021, 11, 22623. [Google Scholar] [CrossRef] [PubMed]
- Tschan, V.J.; Borgna, F.; Schibli, R.; Müller, C. Impact of the Mouse Model and Molar Amount of Injected Ligand on the Tissue Distribution Profile of PSMA Radioligands. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 470–480. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Description | Decay Corrected Activity (%) |
|---|---|---|
| 20 | Starting Activity | 100 |
| 20 | Remnant in PFA | 3.02 ± 0.769 |
| 21 | Pass-through solution | 0.134 ± 0.036 |
| 21 | Loaded DGA | 92.0 ± 0.954 |
| 22 | 5 M HNO3 Wash | 0.051 ± 0.017 |
| 23 | 1 M HCl Wash | 2.90 ± 0.594 |
| 24 | Elution Aliquot #1 | 0.246 ± 0.063 |
| Elution Aliquot #2 | 3.51 ± 1.80 | |
| Elution Aliquot #3 | 10.4 ± 3.33 | |
| Elution Aliquot #4 | 13.7 ± 1.71 | |
| Elution Aliquot #5 | 17.0 ± 1.66 | |
| Elution Aliquot #6 | 13.5 ± 1.50 | |
| Elution Aliquot #7 | 10.5 ± 1.37 | |
| Elution Aliquot #8 | 7.13 ± 1.17 | |
| Elution Aliquot #9 | 4.44 ± 1.00 | |
| Elution Aliquot #10 | 2.72 ± 0.726 | |
| 25 | Remnant in DGA | 3.80 ± 1.17 |
| Total Activity Loss | 9.91 ± 2.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier, J.P.; Bhuiyan, M.; Freifelder, R.; Zhang, H.J.; Gonzalez, L.; Pusateri, A.; Tsai, H.-M.; Leoni, L.; Ghosh, K.; Markiewicz, E.; et al. Synthesis of DOTA-Based 43Sc Radiopharmaceuticals Using Cyclotron-Produced 43Sc as Exemplified by [43Sc]Sc-PSMA-617 for PSMA PET Imaging. Methods Protoc. 2025, 8, 58. https://doi.org/10.3390/mps8030058
Meier JP, Bhuiyan M, Freifelder R, Zhang HJ, Gonzalez L, Pusateri A, Tsai H-M, Leoni L, Ghosh K, Markiewicz E, et al. Synthesis of DOTA-Based 43Sc Radiopharmaceuticals Using Cyclotron-Produced 43Sc as Exemplified by [43Sc]Sc-PSMA-617 for PSMA PET Imaging. Methods and Protocols. 2025; 8(3):58. https://doi.org/10.3390/mps8030058
Chicago/Turabian StyleMeier, Jason P., Mohammed Bhuiyan, Richard Freifelder, Hannah J. Zhang, Lucas Gonzalez, Antonino Pusateri, Hsiu-Ming Tsai, Lara Leoni, Kaustab Ghosh, Erica Markiewicz, and et al. 2025. "Synthesis of DOTA-Based 43Sc Radiopharmaceuticals Using Cyclotron-Produced 43Sc as Exemplified by [43Sc]Sc-PSMA-617 for PSMA PET Imaging" Methods and Protocols 8, no. 3: 58. https://doi.org/10.3390/mps8030058
APA StyleMeier, J. P., Bhuiyan, M., Freifelder, R., Zhang, H. J., Gonzalez, L., Pusateri, A., Tsai, H.-M., Leoni, L., Ghosh, K., Markiewicz, E., Henning, C., Zhang, Y., Weichselbaum, R., Nolen, J., Rotsch, D. A., Kao, C.-M., Szmulewitz, R. Z., Chen, C.-T., & Chitneni, S. K. (2025). Synthesis of DOTA-Based 43Sc Radiopharmaceuticals Using Cyclotron-Produced 43Sc as Exemplified by [43Sc]Sc-PSMA-617 for PSMA PET Imaging. Methods and Protocols, 8(3), 58. https://doi.org/10.3390/mps8030058