
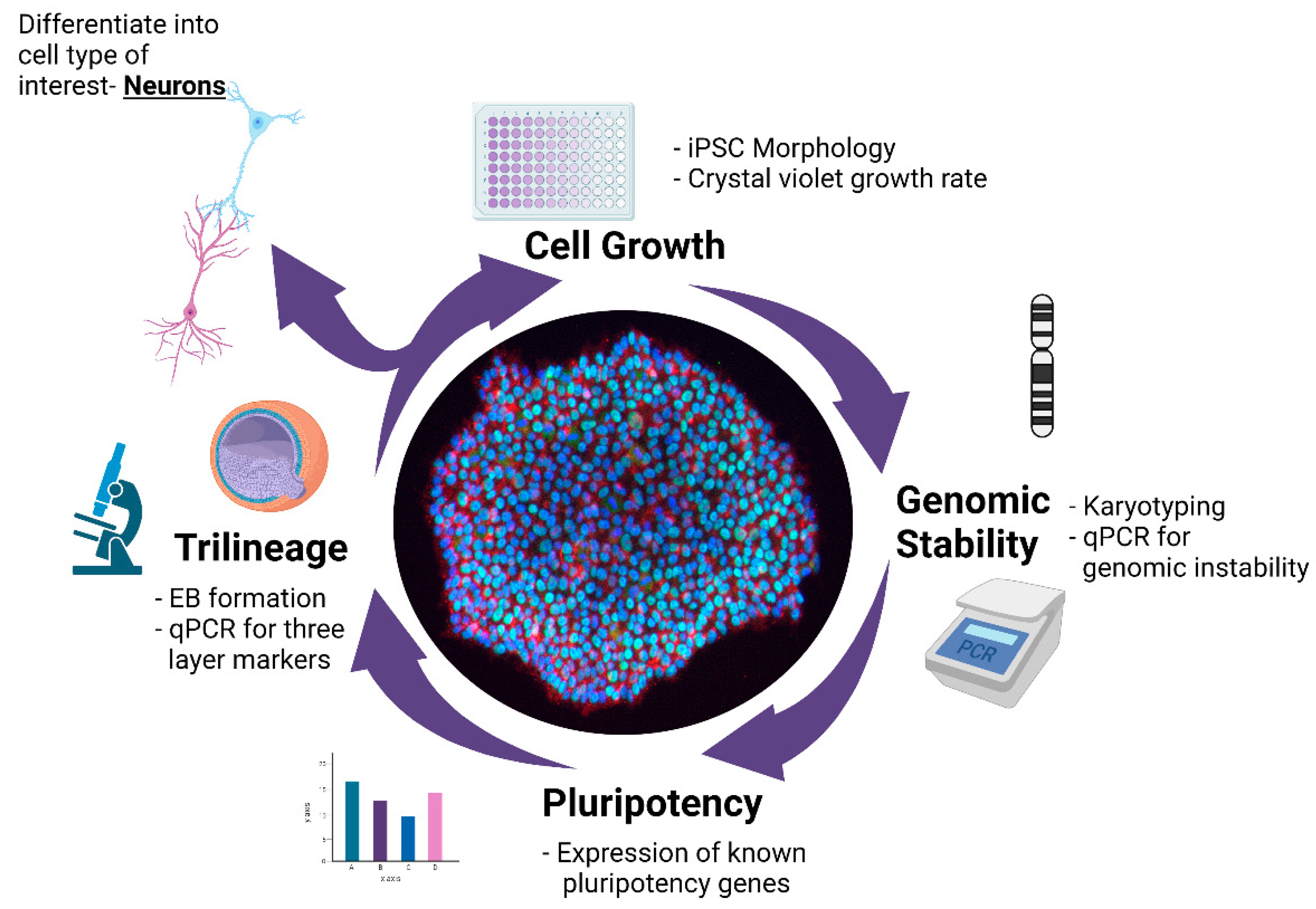
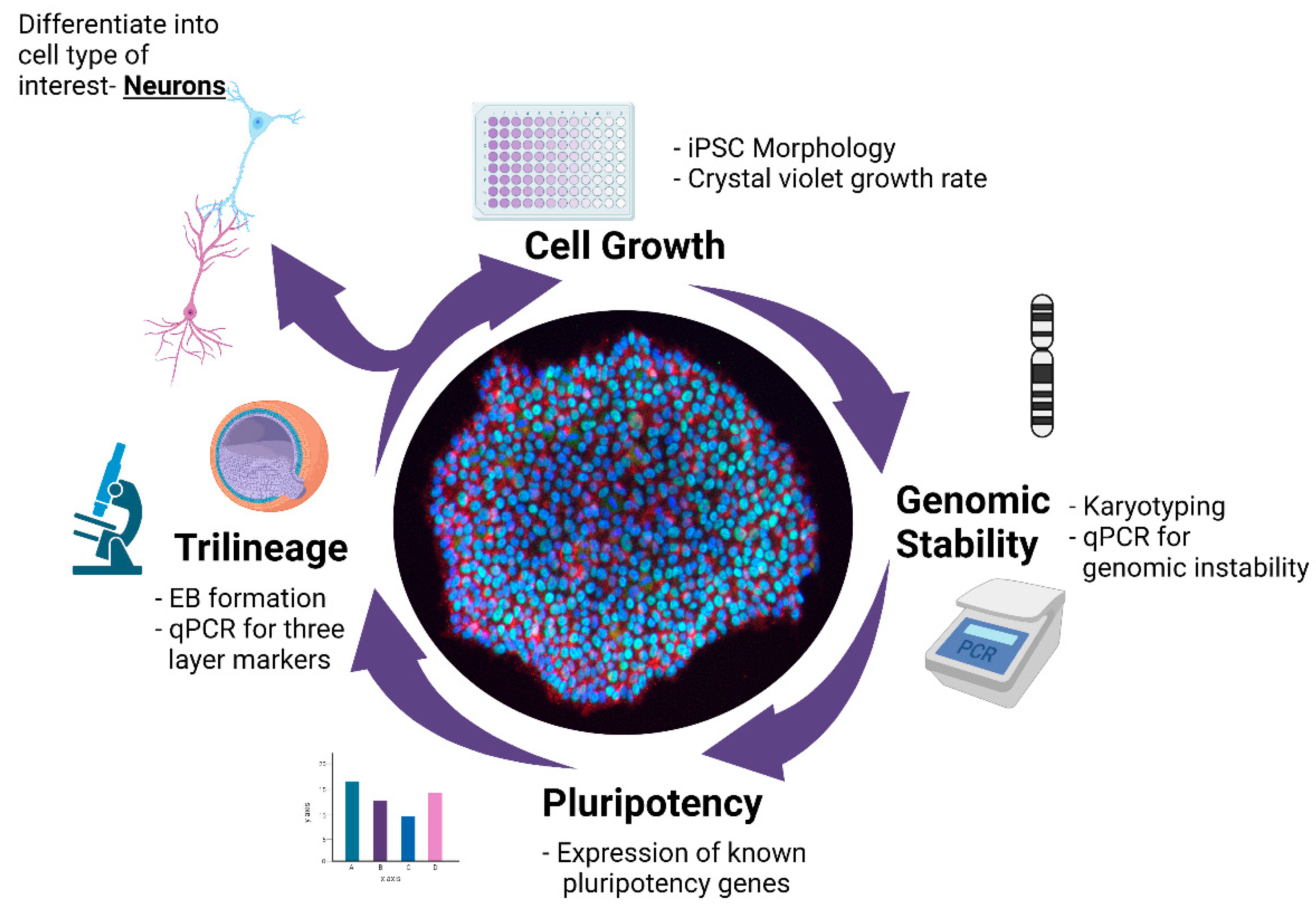
A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. iPSC Reprogramming
2.3. Culture Conditions for iPSCs
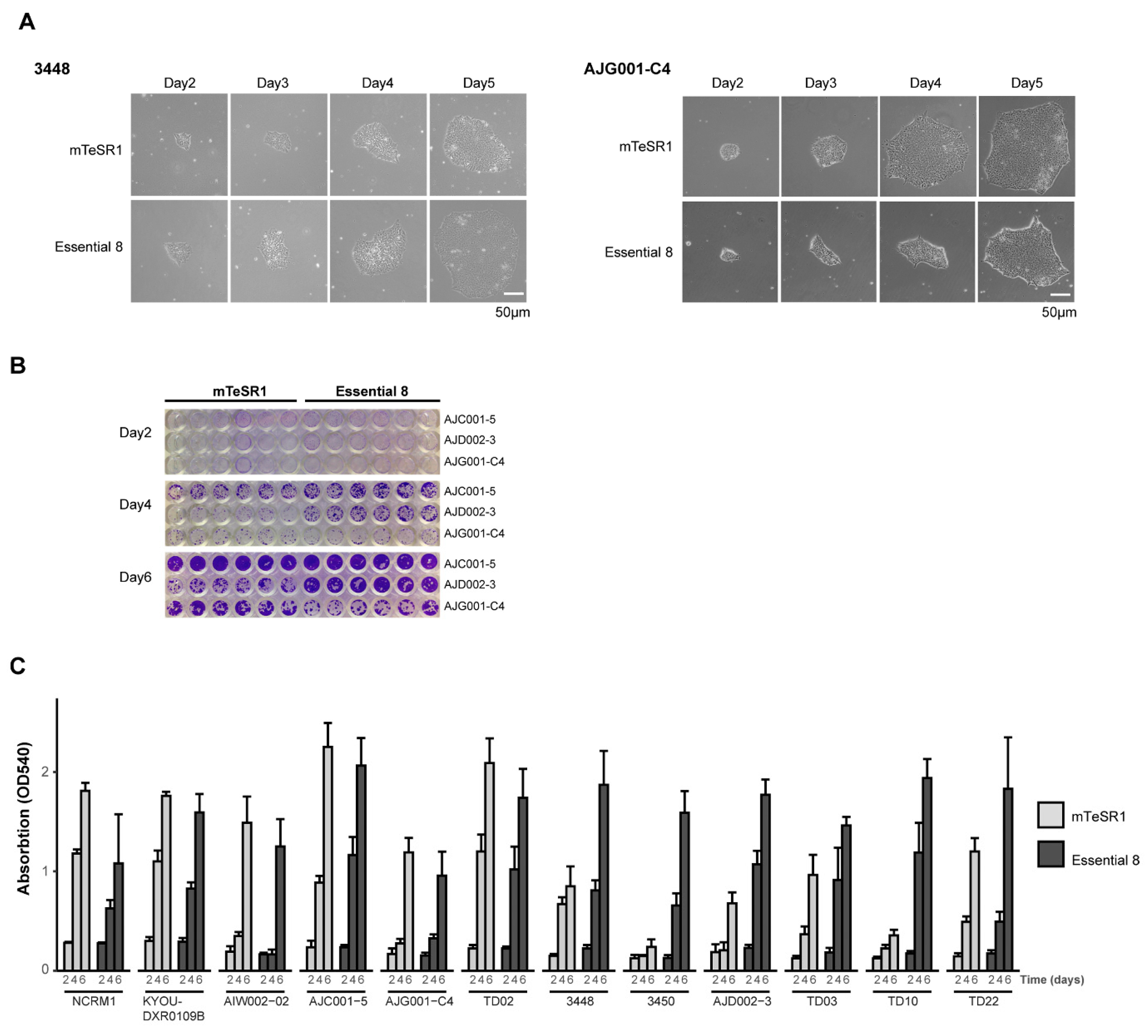
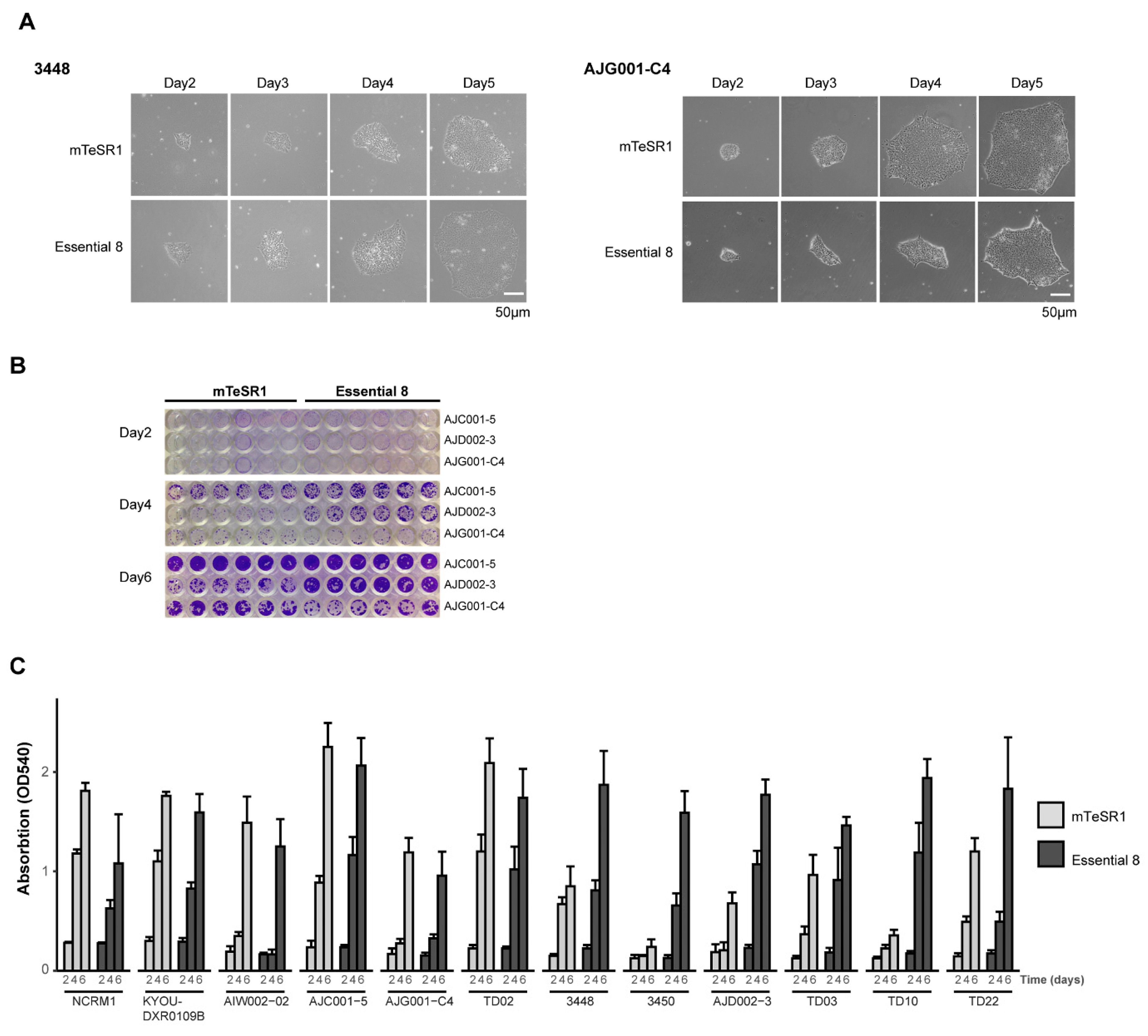
2.4. Crystal Violet Assay
2.5. RNA Isolation, cDNA Synthesis, and qPCR Analysis
2.6. Short Tandem Repeat (STR) Analysis
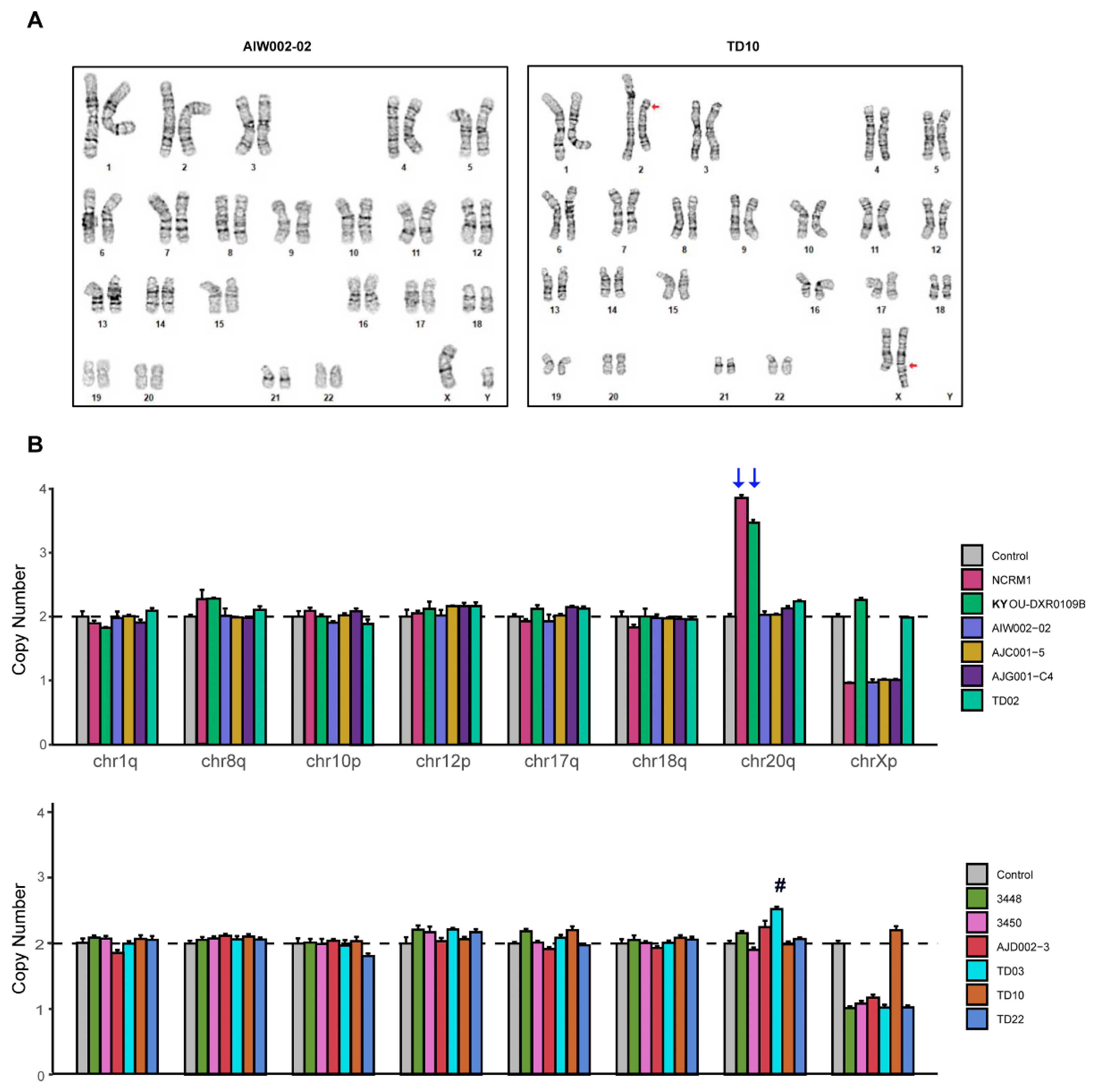
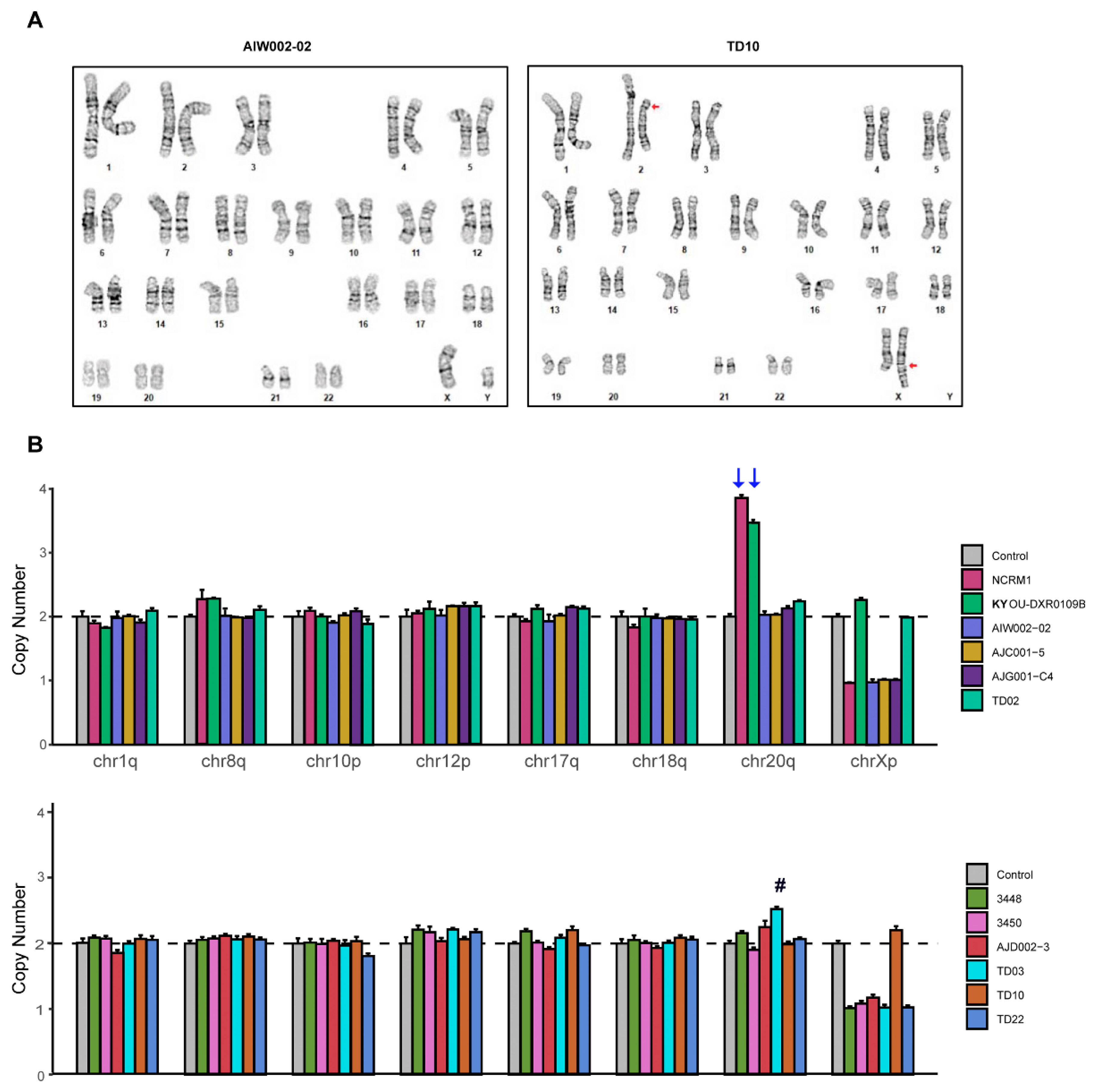
2.7. Karyotyping and Genomic Abnormalities Analysis
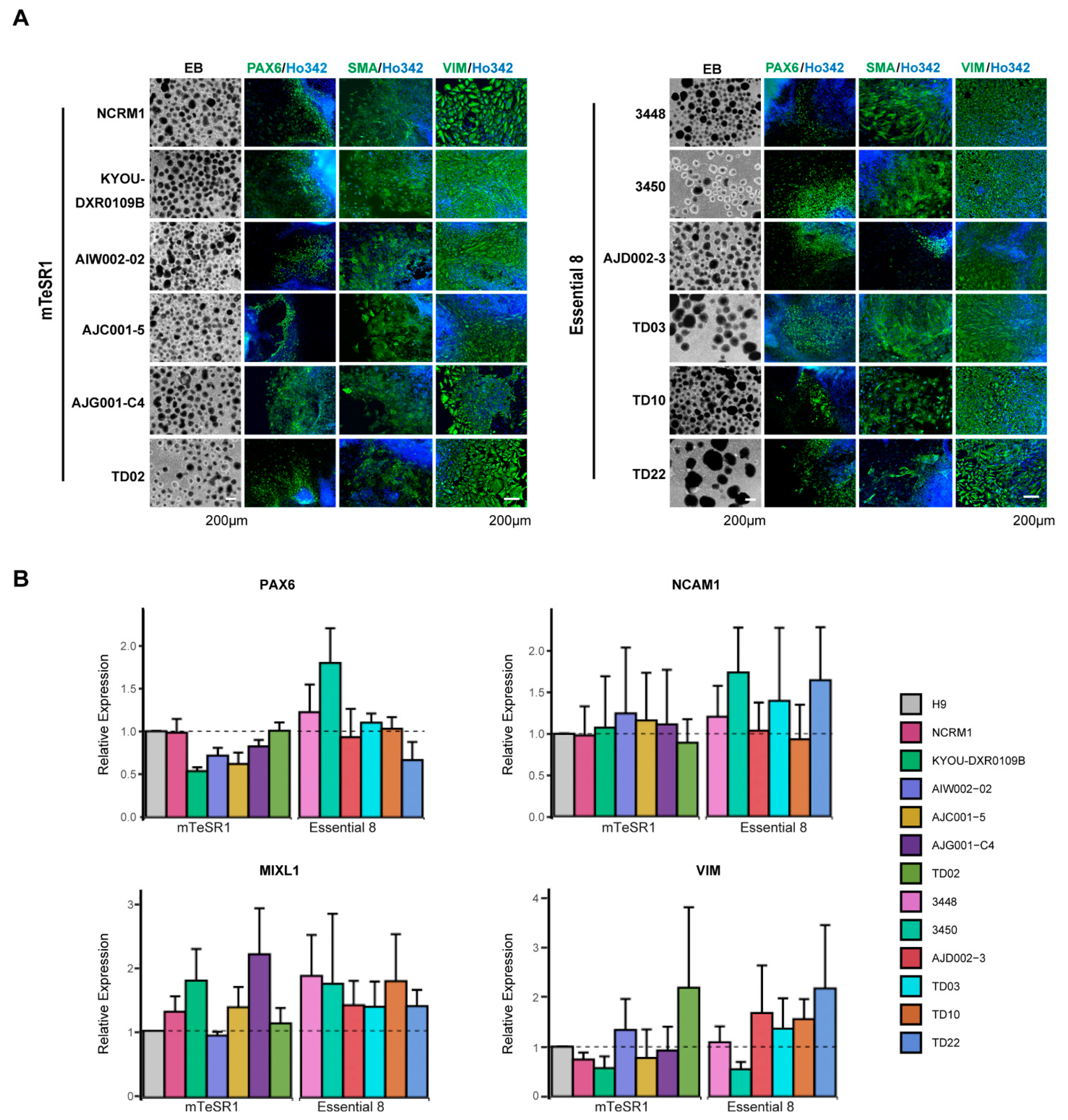
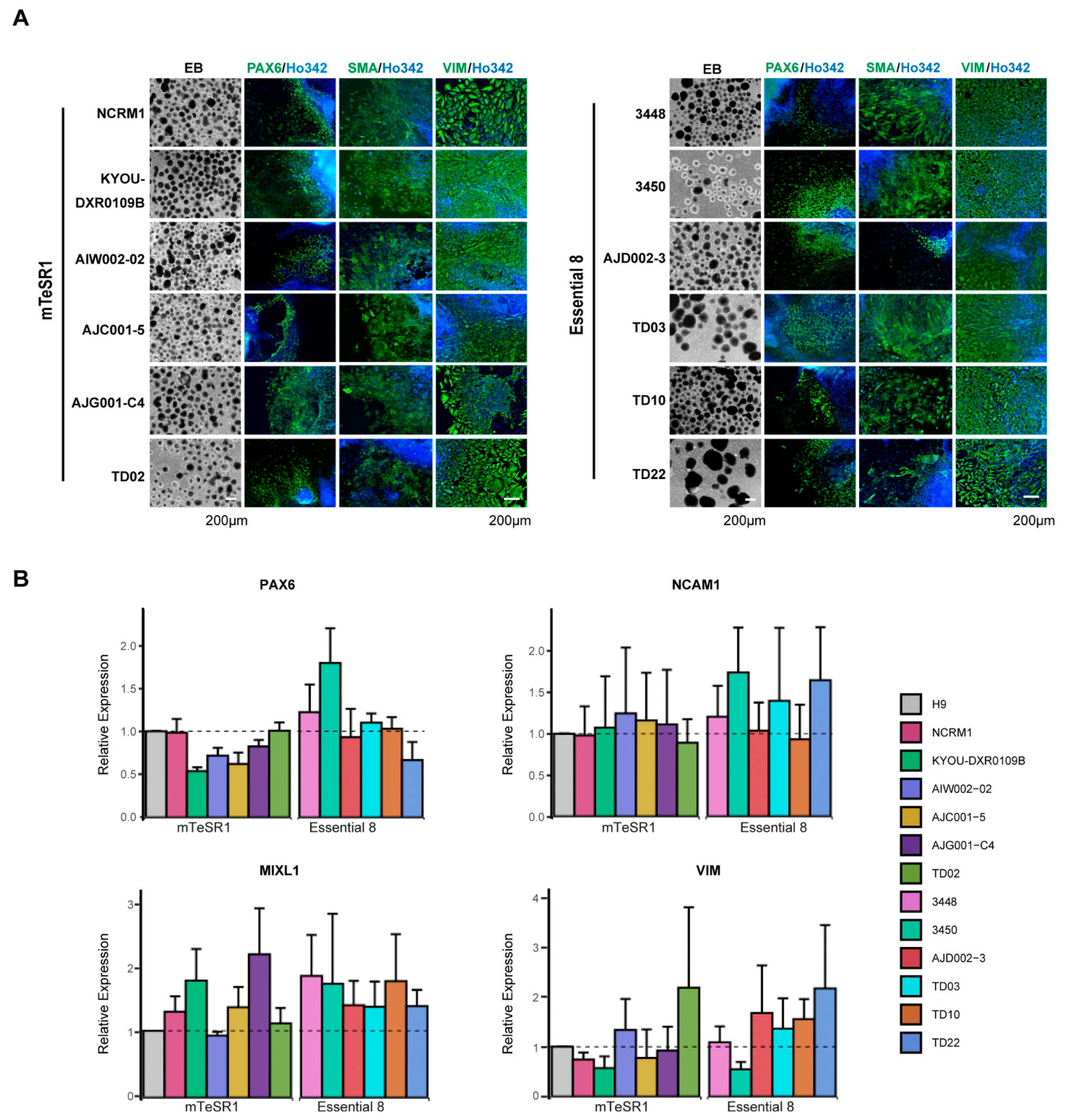
2.8. Three Germ Layer Differentiation Test
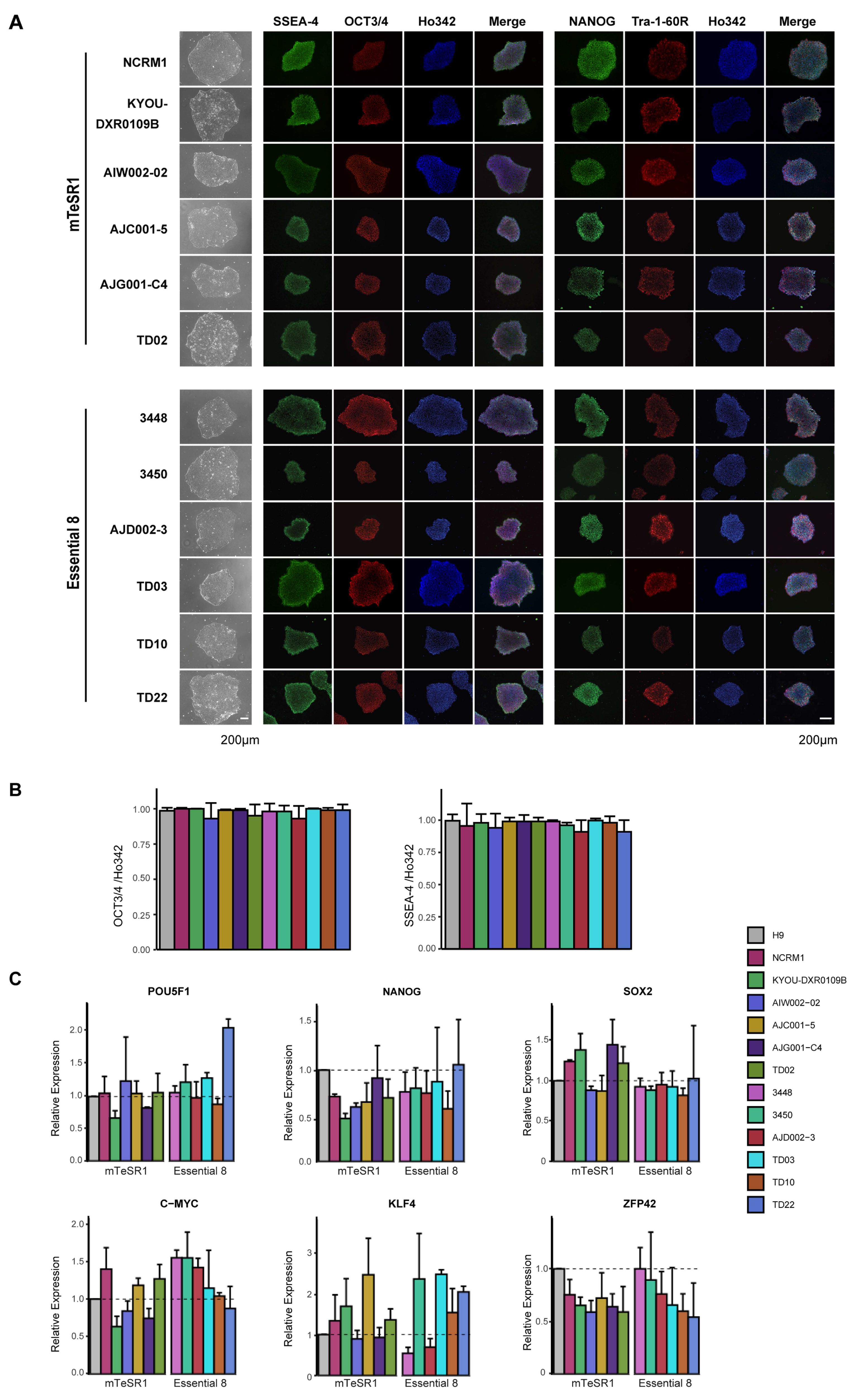
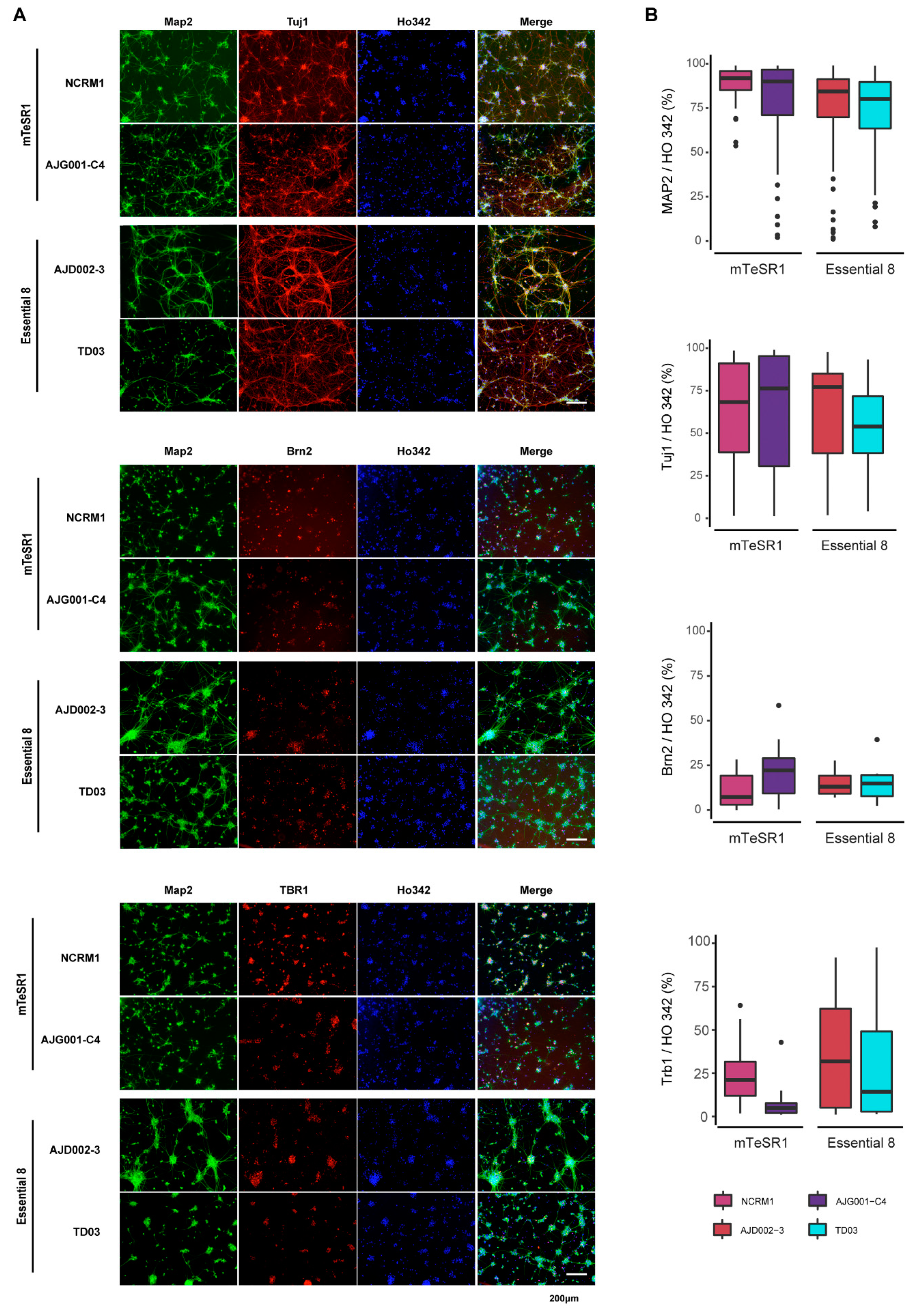
2.9. Immunocytochemistry Analysis
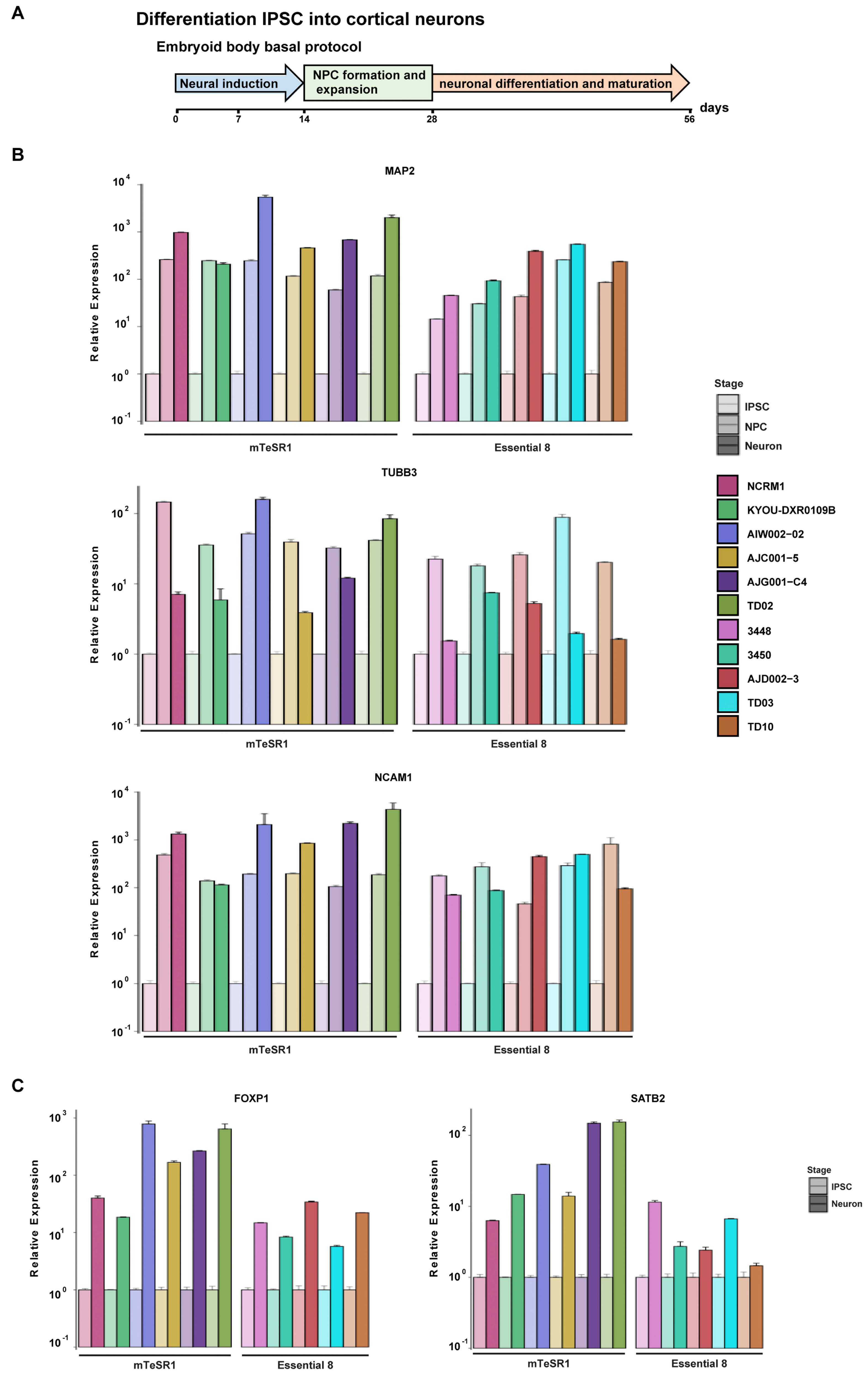
2.10. Cortical Neuron Differentiation
2.11. Data Visualization and Statistical Analysis
3. Results
3.1. Validation of hiPSC Culture Conditions
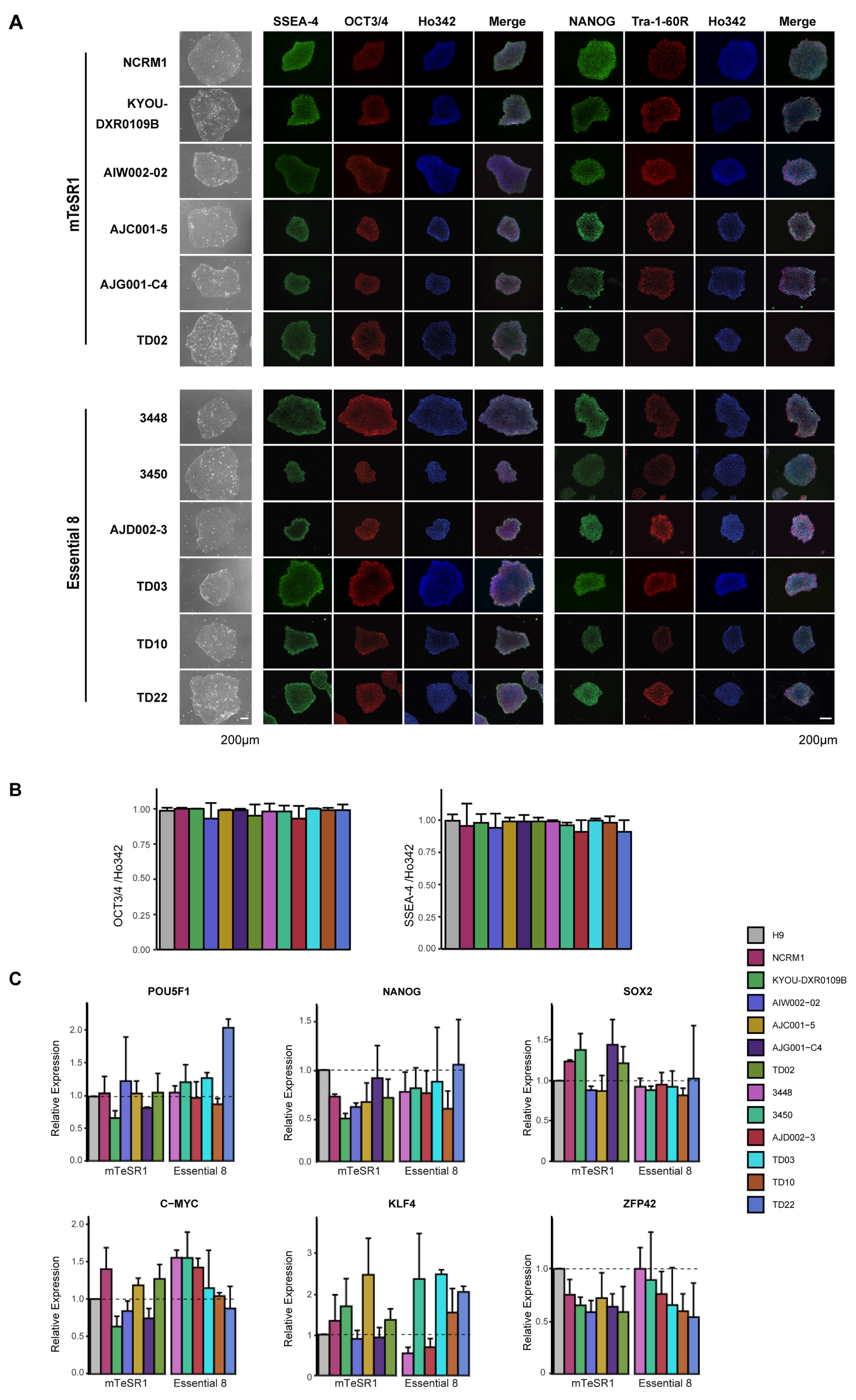
3.2. Characterization of iPSC Pluripotency
3.3. STR and Genomic Abnormality Testing of hiPSCs
3.4. Differentiation of hiPSCs into Three Germ Layers
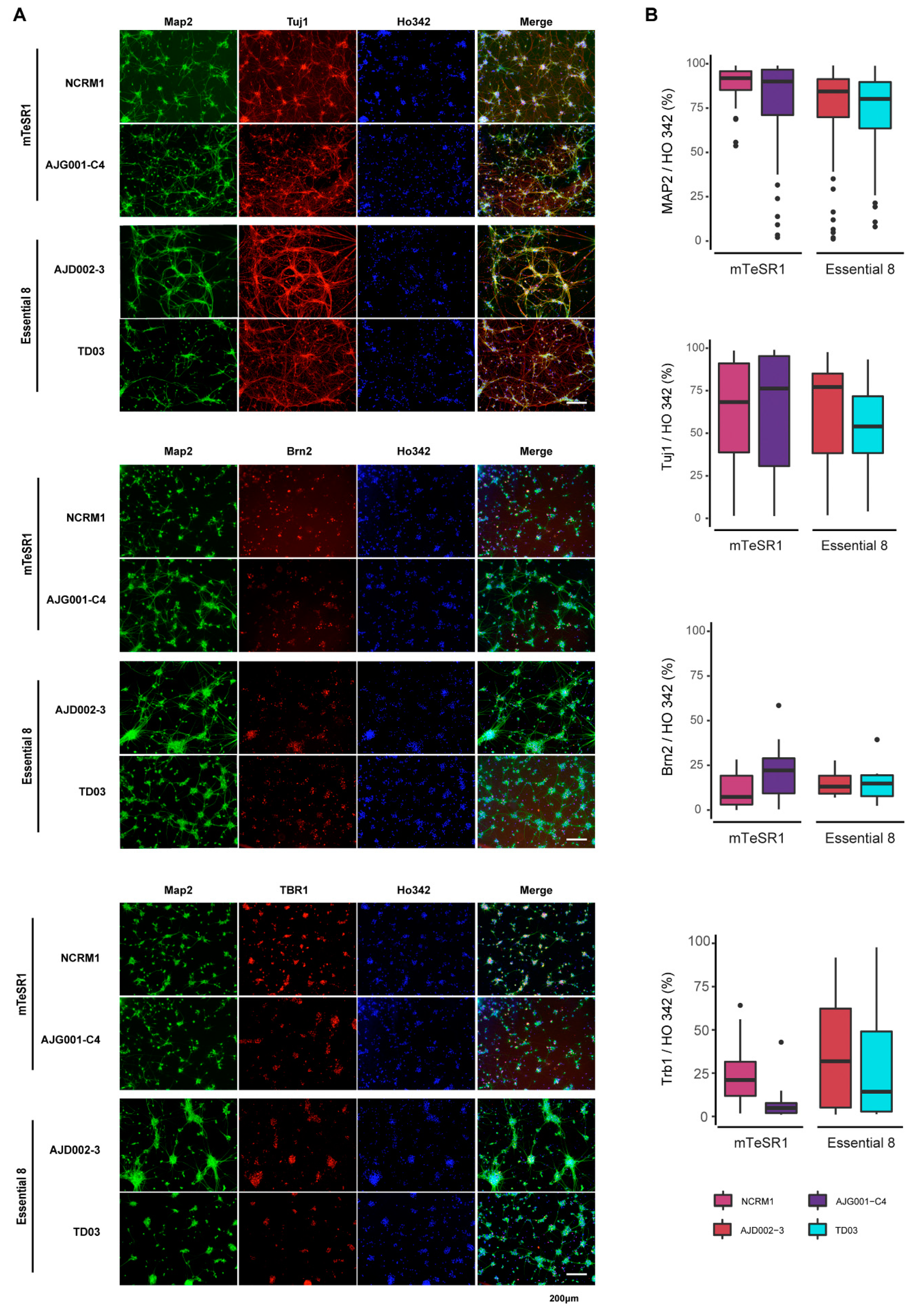
3.5. Differentiation of iPSCs into Cortical Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FAM | 5-carboxyfluorescein |
| CV | crystal violet |
| E8 | Essential 8 media |
| ESC | embryoid body |
| ESC | embryonic stem cell |
| hiPSC | human iPSC |
| Ho342 | Hoechst 33342 |
| ICC | immunocytochemistry |
| iPSC | induced pluripotent stem cell |
| NE | neuroepithelial |
| NEAA | non-essential amino acids |
| NPC | neural progenitor cell |
| PBMC | peripheral blood mononuclear cells |
| PFA | paraformaldehyde |
| QC | quality control |
| RT | room temperature |
References
- Daley, G.Q. The promise and perils of stem cell therapeutics. Cell Stem Cell 2012, 10, 740–749. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Putting stem cells to the test. Nat. Med. 2010, 16, 1354–1357. [Google Scholar] [CrossRef]
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Bennaceur Griscelli, A.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen. Med. 2018, 13, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, T.E.; Levenstein, M.E.; Jones, J.M.; Berggren, W.T.; Mitchen, E.R.; Frane, J.L.; Crandall, L.J.; Daigh, C.A.; Conard, K.R.; Piekarczyk, M.S.; et al. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 2006, 24, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Hey, C.A.B.; Saltokova, K.B.; Bisgaard, H.C.; Moller, L.B. Comparison of two different culture conditions for derivation of early hiPSC. Cell Biol. Int. 2018, 42, 1467–1473. [Google Scholar] [CrossRef]
- Tano, K.; Yasuda, S.; Kuroda, T.; Saito, H.; Umezawa, A.; Sato, Y. A novel in vitro method for detecting undifferentiated human pluripotent stem cells as impurities in cell therapy products using a highly efficient culture system. PLoS ONE 2014, 9, e110496. [Google Scholar] [CrossRef]
- International Stem Cell Initiative, C.; Akopian, V.; Andrews, P.W.; Beil, S.; Benvenisty, N.; Brehm, J.; Christie, M.; Ford, A.; Fox, V.; Gokhale, P.J.; et al. Comparison of defined culture systems for feeder cell free propagation of human embryonic stem cells. In Vitro Cell. Dev. Biol. Anim. 2010, 46, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Ramirez, J.M.; Becker, F.; Pantesco, V.; Lavabre-Bertrand, T.; Hovatta, O.; Lemaitre, J.M.; Pellestor, F.; De Vos, J. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev. 2015, 24, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Grishin, D.; Wang, G.; Aach, J.; Zhang, C.Z.; Chari, R.; Homsy, J.; Cai, X.; Zhao, Y.; Fan, J.B.; et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 2014, 5, 5507. [Google Scholar] [CrossRef]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.E.; Westra, J.W.; Rehen, S.K.; Young, H.; Bushman, D.M.; Paczkowski, C.M.; Yung, Y.C.; Lynch, C.L.; Tran, H.T.; Nickey, K.S.; et al. Normal human pluripotent stem cell lines exhibit pervasive mosaic aneuploidy. PLoS ONE 2011, 6, e23018. [Google Scholar] [CrossRef]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef]
- Martin, U. Genome stability of programmed stem cell products. Adv. Drug Deliv. Rev. 2017, 120, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Mayshar, Y.; Benvenisty, N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell 2011, 9, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Zhang, J.P.; Xu, J.; Su, R.J.; Neises, A.; Ji, G.Z.; Yuan, W.; Cheng, T.; Zhang, X.B. Enhanced generation of integration-free iPSCs from human adult peripheral blood mononuclear cells with an optimal combination of episomal vectors. Stem Cell Rep. 2016, 6, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087379. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Hirst, A.J.; Gokhale, P.J.; Juarez, M.A.; Williams, S.; Wheeler, M.; Bean, K.; Allison, T.F.; Moore, H.D.; Andrews, P.W.; et al. Detecting genetic mosaicism in cultures of human pluripotent stem cells. Stem Cell Rep. 2016, 7, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brustle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Allegrucci, C.; Young, L.E. Differences between human embryonic stem cell lines. Hum. Reprod. Update 2007, 13, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Son, M.Y.; Choi, H.; Han, Y.M.; Cho, Y.S. Unveiling the critical role of REX1 in the regulation of human stem cell pluripotency. Stem Cells 2013, 31, 2374–2387. [Google Scholar] [CrossRef]
- Cabrera, C.M.; Cobo, F.; Nieto, A.; Cortes, J.L.; Montes, R.M.; Catalina, P.; Concha, A. Identity tests: Determination of cell line cross-contamination. Cytotechnology 2006, 51, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, R. Cell biology. Cases of mistaken identity. Science 2007, 315, 928–931. [Google Scholar] [CrossRef]
- Yaffe, M.P.; Noggle, S.A.; Solomon, S.L. Raising the standards of stem cell line quality. Nat. Cell Biol. 2016, 18, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; Beil, S.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, R.J.; Narva, E.; Lahesmaa, R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012, 13, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Narva, E.; Ng, S.; Sourour, M.; Hamalainen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef]
- Narva, E.; Autio, R.; Rahkonen, N.; Kong, L.; Harrison, N.; Kitsberg, D.; Borghese, L.; Itskovitz-Eldor, J.; Rasool, O.; Dvorak, P.; et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat. Biotechnol. 2010, 28, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.S.; Smith, K.; Gokhale, P.; Moore, H.D.; Maltby, E.; Johnson, J.; Meisner, L.; Zwaka, T.P.; Thomson, J.A.; Andrews, P.W. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat. Biotechnol. 2004, 22, 53–54. [Google Scholar] [CrossRef]
- Inzunza, J.; Sahlen, S.; Holmberg, K.; Stromberg, A.M.; Teerijoki, H.; Blennow, E.; Hovatta, O.; Malmgren, H. Comparative genomic hybridization and karyotyping of human embryonic stem cells reveals the occurrence of an isodicentric X chromosome after long-term cultivation. Mol. Hum. Reprod. 2004, 10, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Lefort, N.; Feyeux, M.; Bas, C.; Feraud, O.; Bennaceur-Griscelli, A.; Tachdjian, G.; Peschanski, M.; Perrier, A.L. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat. Biotechnol. 2008, 26, 1364–1366. [Google Scholar] [CrossRef]
- Sareen, D.; McMillan, E.; Ebert, A.D.; Shelley, B.C.; Johnson, J.A.; Meisner, L.F.; Svendsen, C.N. Chromosome 7 and 19 trisomy in cultured human neural progenitor cells. PLoS ONE 2009, 4, e7630. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, M.; Martins-Taylor, K.; Wang, X.; Zhang, Z.; Park, J.W.; Zhan, S.; Kronenberg, M.S.; Lichtler, A.; Liu, H.X.; et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS ONE 2010, 5, e11853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engle, S.J.; Blaha, L.; Kleiman, R.J. Best Practices for Translational Disease Modeling Using Human iPSC-Derived Neurons. Neuron 2018, 100, 783–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamm, N.; Ben-David, U.; Golan-Lev, T.; Storchova, Z.; Benvenisty, N.; Kerem, B. Genomic Instability in Human Pluripotent Stem Cells Arises from Replicative Stress and Chromosome Condensation Defects. Cell Stem Cell 2016, 18, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, S.; Lopez-Contreras, A.J.; Gabut, M.; Marion, R.M.; Gutierrez-Martinez, P.; Bua, S.; Ramirez, O.; Olalde, I.; Rodrigo-Perez, S.; Li, H.; et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat. Commun. 2015, 6, 8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, J.; Skylaki, S.; Iwabuchi, K.A.; Chantzoura, E.; Ruetz, T.; Johnsson, A.; Tomlinson, S.R.; Linnarsson, S.; Kaji, K. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature 2013, 499, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Papapetrou, E.P.; Tomishima, M.J.; Chambers, S.M.; Mica, Y.; Reed, E.; Menon, J.; Tabar, V.; Mo, Q.; Studer, L.; Sadelain, M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 12759–12764. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Hirano, K.; Nagata, S.; Tada, T. Sox2 expression effects on direct reprogramming efficiency as determined by alternative somatic cell fate. Stem Cell Res. 2011, 6, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Matoba, R.; Niwa, H.; Masui, S.; Ohtsuka, S.; Carter, M.G.; Sharov, A.A.; Ko, M.S. Dissecting Oct3/4-regulated gene networks in embryonic stem cells by expression profiling. PLoS ONE 2006, 1, e26. [Google Scholar] [CrossRef]
- Daniszewski, M.; Nguyen, Q.; Chy, H.S.; Singh, V.; Crombie, D.E.; Kulkarni, T.; Liang, H.H.; Sivakumaran, P.; Lidgerwood, G.E.; Hernandez, D.; et al. Single-cell profiling identifies key pathways expressed by iPSCs cultured in different commercial media. iScience 2018, 7, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.H.; Lukowski, S.W.; Chiu, H.S.; Senabouth, A.; Bruxner, T.J.C.; Christ, A.N.; Palpant, N.J.; Powell, J.E. Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018, 28, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Peterson, S.E.; Tran, H.T.; Garitaonandia, I.; Han, S.; Nickey, K.S.; Leonardo, T.; Laurent, L.C.; Loring, J.F. Teratoma generation in the testis capsule. J. Vis. Exp. 2011, e3177. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.J.; Brändl, B.; Loring, J.F. Assessment of human pluripotent stem cells with PluriTest. Int. J. Biomed. Health Sci. 2013, 9, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Ungrin, M.D.; Joshi, C.; Nica, A.; Bauwens, C.; Zandstra, P.W. Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLoS ONE 2008, 3, e1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Zhang, Y. Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell Stem Cell 2013, 13, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic instability of iPSCs: Challenges towards their clinical applications. Stem Cell Rev. Rep. 2017, 13, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.H.; Sampsell-Barron, T.L.; Gu, F.; Root, S.; Peck, R.M.; Pan, G.; Yu, J.; Antosiewicz-Bourget, J.; Tian, S.; Stewart, R.; et al. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 2008, 3, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Chen, X.; Li, D.S.; Li, R.; Addicks, G.C.; Glennon, C.; Zwaka, T.P.; Thomson, J.A. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol. 2002, 20, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Maussion, G.; Diallo, A.B.; Gigek, C.O.; Chen, E.S.; Crapper, L.; Théroux, J.F.; Chen, G.G.; Vasuta, C.; Ernst, C. Investigation of genes important in neurodevelopment disorders in adult human brain. Hum. Genet. 2015, 134, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Sugitani, Y.; Nakai, S.; Minowa, O.; Nishi, M.; Jishage, K.; Kawano, H.; Mori, K.; Ogawa, M.; Noda, T. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 2002, 16, 1760–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britanova, O.; de Juan Romero, C.; Cheung, A.; Kwan, K.Y.; Schwark, M.; Gyorgy, A.; Vogel, T.; Akopov, S.; Mitkovski, M.; Agoston, D.; et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 2008, 57, 378–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Donor | Cell Type | Donor Age | Sex 1 | Cell Source | Reprogramming | Media 2 |
|---|---|---|---|---|---|---|---|
| H9 | ESC | NA | F | WiCell | mTeSR1 | ||
| NCRM1 | A | cord blood | NA | M | NIH | Episomal | mTeSR1 |
| KYOU-DXR0109B | B | fibroblast | 36 | F | ATCC | Sendai virus | mTeSR1 |
| AIW002-02 | C | PBMC | 37 | M | The Neuro | Sendai virus | mTeSR1 |
| AJC001-5 | C | fibroblast | 37 | M | The Neuro | Sendai virus | mTeSR1 |
| AJG001-C4 | C | PBMC | 37 | M | The Neuro | Episomal | mTeSR1 |
| TD02 | D | PBMC | 48 | F | The Neuro | Episomal | mTeSR1 |
| 3448 | E | PBMC | 48 | M | The Neuro | Episomal | E8 |
| 3450 | F | PBMC | 37 | M | The Neuro | Episomal | E8 |
| AJD002-3 | G | PBMC | 44 | M | The Neuro | Sendai virus | E8 |
| TD03 | G | PBMC | 44 | M | The Neuro | Episomal | E8 |
| TD10 | H | PBMC | 64 | F | The Neuro | Episomal | E8 |
| TD22 | I | PBMC | 59 | M | The Neuro | Episomal | E8 |
| 3059 PBMC | AJD002-3 iPSC | TD03 iPSC | ||||
|---|---|---|---|---|---|---|
| Marker | Allele 1 | Allele 2 | Allele 1 | Allele 2 | Allele 1 | Allele 2 |
| AMEL | X | Y | X | Y | X | Y |
| CSF1PO | 11 | 12 | 11 | 12 | 11 | 12 |
| D13S317 | 9 | 11 | 9 | 11 | 9 | 11 |
| D16S539 | 9 | 11 | 9 | 11 | 9 | 11 |
| D21S11 | 28 | 30 | 28 | 30 | 28 | 30 |
| D5S818 | 9 | 11 | 9 | 11 | 9 | 11 |
| D7S820 | 12 | 12 | 12 | 12 | 12 | 12 |
| TH01 | 6 | 9 | 6 | 9 | 6 | 9 |
| TPOX | 8 | 9 | 8 | 9 | 8 | 9 |
| vWA | 14 | 15 | 14 | 15 | 14 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.X.-Q.; Abdian, N.; Maussion, G.; Thomas, R.A.; Demirova, I.; Cai, E.; Tabatabaei, M.; Beitel, L.K.; Karamchandani, J.; Fon, E.A.; et al. A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types. Methods Protoc. 2021, 4, 50. https://doi.org/10.3390/mps4030050
Chen CX-Q, Abdian N, Maussion G, Thomas RA, Demirova I, Cai E, Tabatabaei M, Beitel LK, Karamchandani J, Fon EA, et al. A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types. Methods and Protocols. 2021; 4(3):50. https://doi.org/10.3390/mps4030050
Chicago/Turabian StyleChen, Carol X.-Q., Narges Abdian, Gilles Maussion, Rhalena A. Thomas, Iveta Demirova, Eddie Cai, Mahdieh Tabatabaei, Lenore K. Beitel, Jason Karamchandani, Edward A. Fon, and et al. 2021. "A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types" Methods and Protocols 4, no. 3: 50. https://doi.org/10.3390/mps4030050
APA StyleChen, C. X.-Q., Abdian, N., Maussion, G., Thomas, R. A., Demirova, I., Cai, E., Tabatabaei, M., Beitel, L. K., Karamchandani, J., Fon, E. A., & Durcan, T. M. (2021). A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types. Methods and Protocols, 4(3), 50. https://doi.org/10.3390/mps4030050