miRNA Targets: From Prediction Tools to Experimental Validation




Abstract
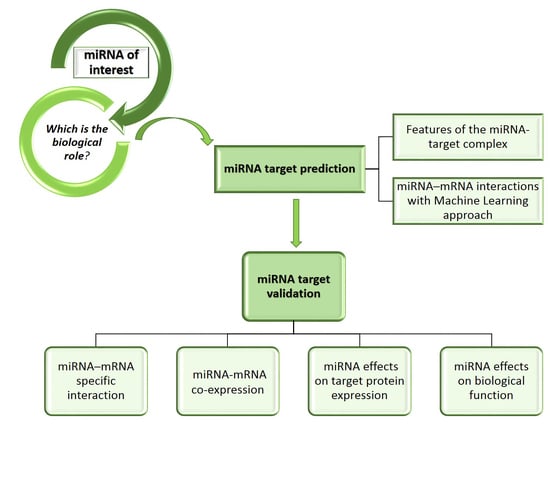
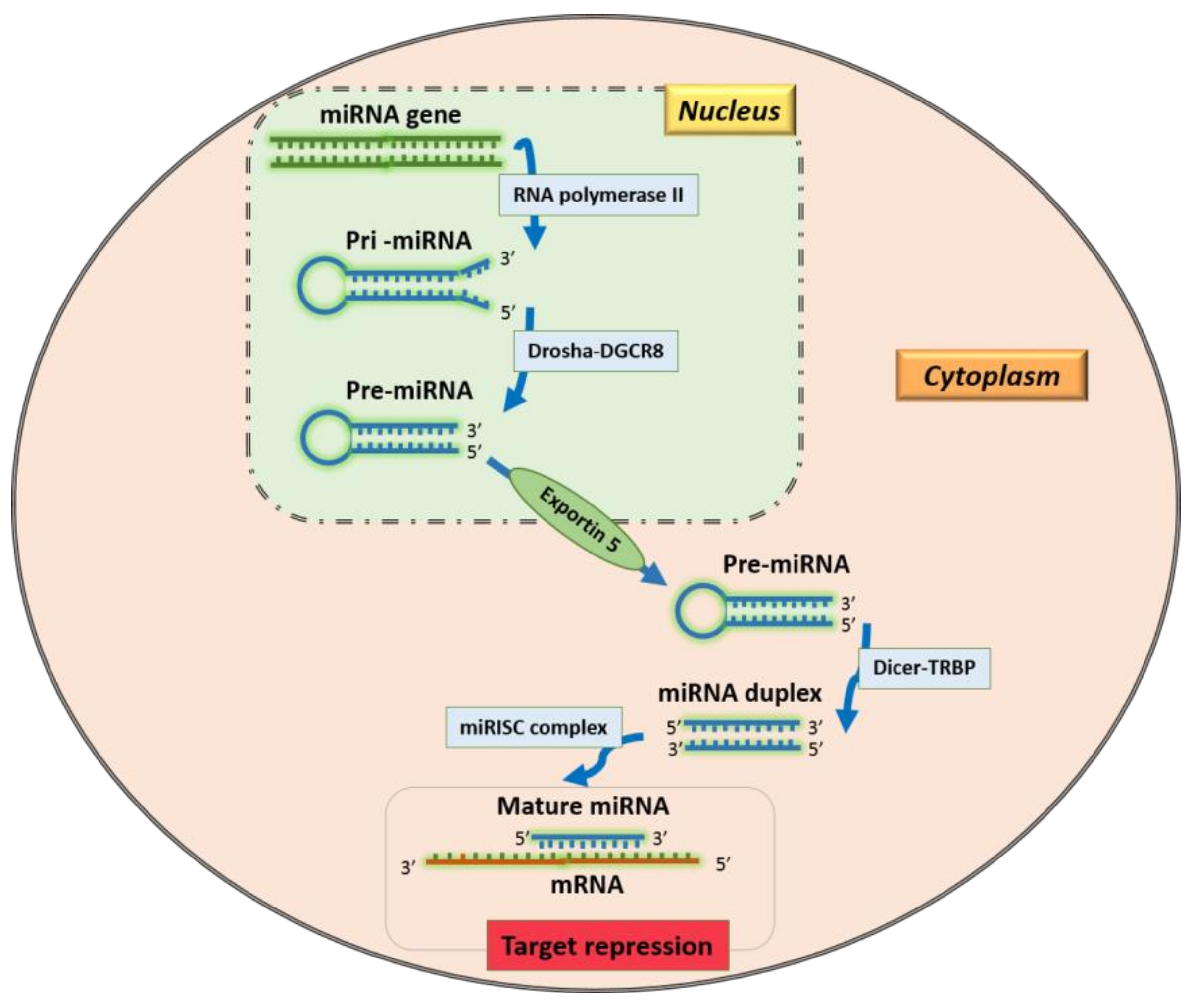
1. Introduction
2. miRNA Target Prediction
2.1. Predictive Methods
2.1.1. Strategies for de novo Predictions
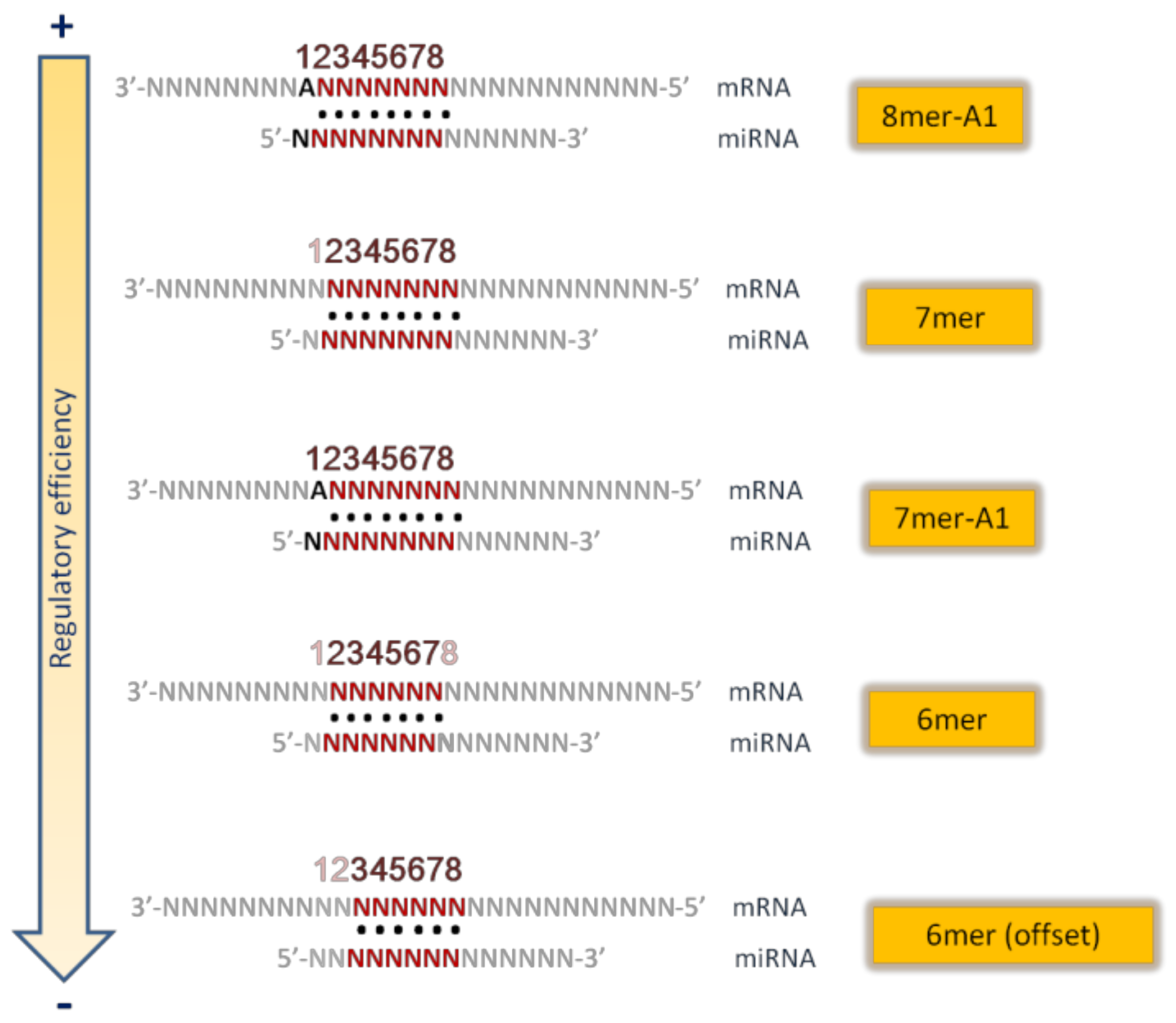
- Seed pairing
- 8mer site: a perfect Watson-Crick match from nucleotide 2 to nucleotide 8 of the miRNA seed, with an “A” in mRNA opposite position 1;
- 7mer site: Watson-Crick match from nucleotide 2 to nucleotide 8 without the “A” opposite position 1;
- 7mer A1 site: Watson–Crick match from nucleotide 2 to nucleotide 7 with an A opposite position 1;
- 6mer site: position 2–7 match;
- 6mer site: position 3–8 match.

- 2.
- Thermodynamic stability
- 3.
- Evolutionary conservation
- 4.
- Accessibility of target site
- 5.
- Number of target sites in the same 3′-UTR
2.1.2. Machine Learning
- For every miRNA, identify the presumed binding site from validated mRNA targets (as positive) and non-targets (as negative).
- Extract features from these interactions (regardless of whether they are functional or nonfunctional).
- Train a classifier to discriminate targets from non-targets.
- Test the classifier.
- Use the classifier to sort unknown miRNA-mRNA interactions as positive (target) or negative (non-target).


2.1.3. Operative Strategy

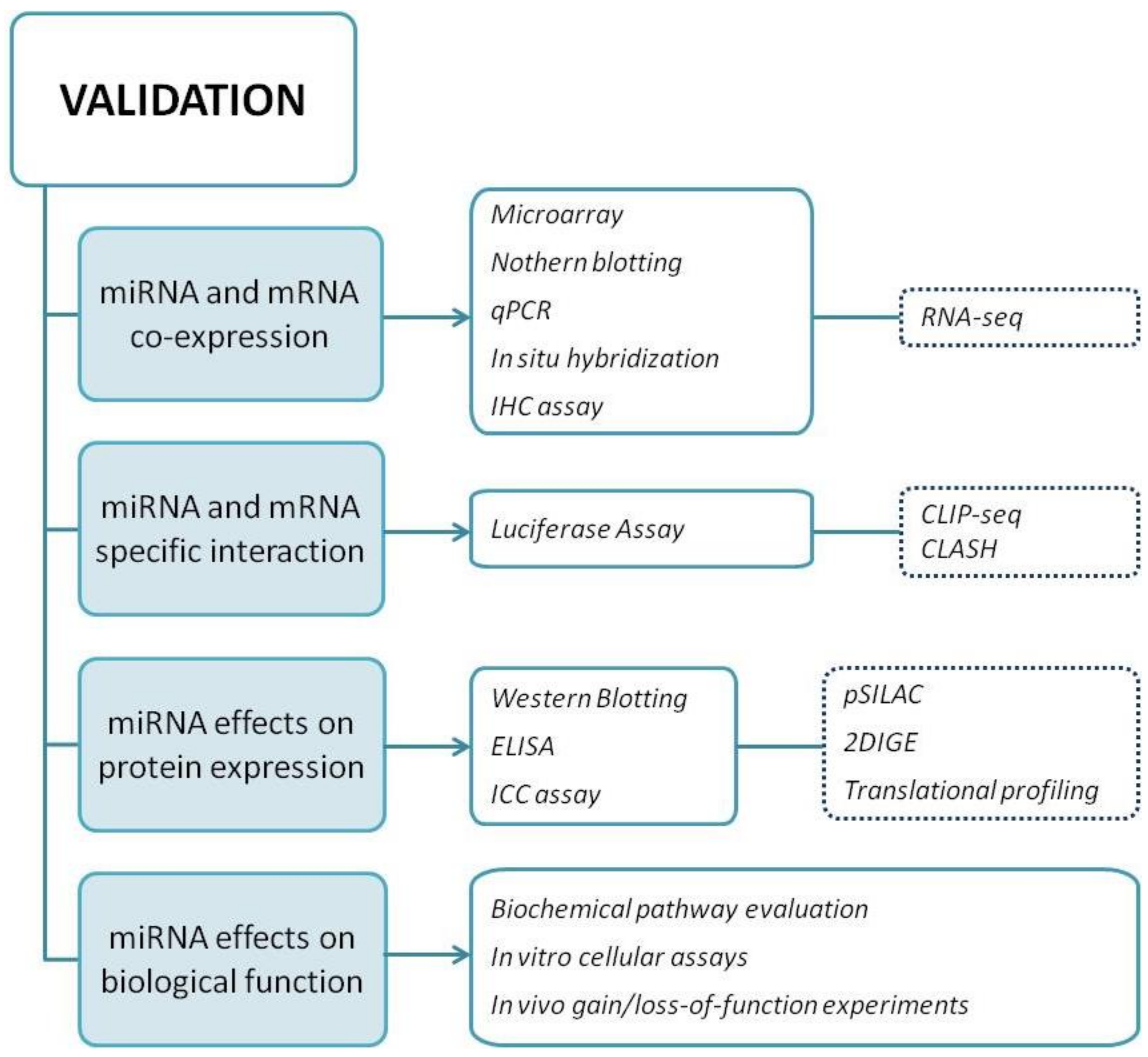
3. miRNA Target Validation
3.1. Validation Methods
3.1.1. Criterion I: Show Co-Expression of miRNA and Target mRNA In Vivo
3.1.2. Criterion II: Prove Interaction between miRNA and a Specific mRE Target Site
3.1.3. Criterion III: Demonstrate miRNA-Mediated Effects on Target Protein Expression
3.1.4. Criterion IV: Demonstrate miRNA Effects on Biological Function
3.2. High-Throughput Technologies
3.2.1. Transcriptomic Analysis and Sequencing
- (1)
- (2)
- (3)
- These assays are time consuming and labour intensive.
- (4)
- Accessibility to mRE sites by miRNA may be very difficult when 3′-UTR sequence gives rise to a complicated secondary structure [84].
- (5)
- The choice of a cell culture as a suitable model to clearly detect consequences of miRNA mimic/inhibitor transfection is challenging: it should express appropriate amounts of endogenous miRNA and target gene.
3.2.2. Biochemical Assays
3.2.3. Proteomic Analysis
3.3. Strengths and Limitations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y. Non-coding RNA. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Stroynowska-Czerwinska, A.; Fiszer, A.; Krzyzosiak, W.J. The panorama of miRNA-mediated mechanisms in mammalian cells. Cell. Mol. Life Sci. 2014, 71, 2253–2270. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Kern, F.; Backes, C.; Hirsch, P.; Fehlmann, T.; Hart, M.; Meese, E.; Keller, A. What’s the target: Understanding two decades of in silico microRNA-target prediction. Brief. Bioinform. 2019, 21, 1999–2010. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning, 1st ed.; Springer: Cambridge, UK, 2006. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Schirle, N.T.; Sheu-Gruttadauria, J.; MacRae, I.J. Structural basis for microRNA targeting. Science 2014, 346, 608–613. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.B.; Shomron, N.; Sandberg, R.; Hornstein, E.; Kitzman, J.; Burge, C.B. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA 2007, 13, 1894–1910. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Micolucci, L.; Islam, M.S.; Olivieri, F.; Procopio, A.D. A practical guide to miRNA target prediction. Methods Mol. Biol. 2019, 1970, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thadani, R.; Tammi, M.T. MicroTar: Predicting microRNA targets from RNA duplexes. BMC Bioinform. 2006, 7, S20. [Google Scholar] [CrossRef] [PubMed]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Yue, D.; Liu, H.; Huang, Y. Survey of computational algorithms for microRNA target prediction. Curr. Genom. 2009, 10, 478–492. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2005, 3, e264, Erratum in 2005, 3, e264. [Google Scholar] [CrossRef]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 2006, 12, 192–197. [Google Scholar] [CrossRef]
- Kobayashi, H.; Tomari, Y. RISC assembly: Coordination between small RNAs and Argonaute proteins. Biochim. Biophys. Acta 2016, 1859, 71–81. [Google Scholar] [CrossRef]
- Robins, H.; Li, Y.; Padgett, R.W. Incorporating structure to predict microRNA targets. Proc. Natl. Acad. Sci. USA 2005, 102, 4006–4009. [Google Scholar] [CrossRef]
- Long, D.; Lee, R.; Williams, P.; Chan, C.Y.; Ambros, V.; Ding, Y. Potent effect of target structure on microRNA function. Nat. Struct. Mol. Biol. 2007, 14, 287. [Google Scholar] [CrossRef] [PubMed]
- Marìn, R.M.; Vanìcek, J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2010, 39, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Yachie, N.; Numata, K.; Saito, R.; Kanai, A.; Tomita, M. Computational analysis of microRNA targets in Caenorhabditis elegans. Gene 2006, 365, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Šulc, M.; Marín, R.M.; Robins, H.S.; Vaníček, J. PACCMIT/PACCMIT-CDS: Identifying microRNA targets in 3′ UTRs and coding sequences. Nucleic Acids Res. 2015, 43, W474–W479. [Google Scholar] [CrossRef]
- Gumienny, R.; Zavolan, M. Accurate transcriptome-wide prediction of microRNA targets and small interfering RNA off-targets with MIRZA-G. Nucleic Acids Res. 2015, 43, 1380–1391. [Google Scholar] [CrossRef]
- Loher, P.; Rigoutsos, I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 2012, 28, 3322–3323. [Google Scholar] [CrossRef] [PubMed]
- Samuel, A.L. Some studies in Machine Learning using the game of checkers. IBM J. Res. Dev. 1959, 3, 210–229. [Google Scholar] [CrossRef]
- Parveen, A.; Mustafa, S.H.; Yadav, P.; Kumar, A. Applications of Machine Learning in miRNA discovery and target prediction. Curr. Genom. 2019, 20, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Shrestha, S.; Yang, C.-D.; Chang, N.-W.; Lin, Y.-L.; Liao, K.-W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA target interactions. Nucleic Acids Res. 2017, 46, D296–D302. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G. DIANA-tarbase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2017, 46, D239–D245. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Schäfer, M.; Ciaudo, C. Prediction of the miRNA interactome—Established methods and upcoming perspectives. Comput. Struct. Biotechnol. J. 2020, 18, 548–557. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Ghosh, D.; Mitra, R.; Zhao, Z. MBSTAR: Multiple instance learning for predicting specific functional binding sites in microRNA targets. Sci. Rep. 2015, 5, 8004. [Google Scholar] [CrossRef]
- Yousef, M.; Jung, S.; Kossenkov, A.V.; Showe, L.C.; Showe, M.K. Naïve Bayes for microRNA target predictions-Machine Learning for microRNA targets. Bioinformatics 2007, 23, 2987–2992. [Google Scholar] [CrossRef]
- Saetrom, O.; Snǿve, O.; Saetrom, P. Weighted sequence motifs as an improved seeding step in microRNA target prediction algorithms. RNA 2005, 11, 995–1003. [Google Scholar] [CrossRef]
- Lee, B.; Baek, J.; Park, S.; Yoon, S. DeepTarget. End-to-end learning framework for microRNA target prediction using deep recurrent neural networks. BCB 2016, 434–442. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mitra, R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics 2009, 25, 2625–2631. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Cong, P.; Zhang, Z.; Lu, H.; Li, T. DeepMirTar: A deep-learning approach for predicting human miRNA targets. Bioinformatics 2018, 34, 3781–3787. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, W169–W173. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Maragkakis, M.; Reczko, M.; Simossis, V.A.; Alexiou, P.; Papadopoulos, G.L.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; et al. DIANA-microT web server: Elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009, 37, W273–W276. [Google Scholar] [CrossRef]
- Farh, K.K.-H.; Grimson, A.; Jan, C.; Lewis, B.P.; Johnston, W.K.; Lim, L.P.; Burge, C.B.; Bartel, D.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science 2005, 310, 1817–1821. [Google Scholar] [CrossRef]
- Borchert, G.M.; Gilmore, B.L.; Spengler, R.M.; Xing, Y.; Lanier, W.; Bhattacharya, D.; Davidson, B.L. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum. Mol. Genet. 2009, 18, 4801–4807. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, H.; Li, L. Alternative mRNA processing increases the complexity of microRNA-based gene regulation in Arabidopsis. Plant. J. 2012, 70, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5-UTR and 3-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef]
- Marin, R.M.; Šulc, M.; Vanicek, J. Searching the coding region for microRNA targets. RNA 2013, 19, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.C.; Bovolenta, L.A.; Nachtigall, P.G.; Herkenhoff, M.E.; Lemke, N.; Pinhal, D. Combining results from distinct microRNA target prediction tools enhances the performance of analyses. Front. Genet. 2017, 8, 59. [Google Scholar] [CrossRef]
- Fan, Q.; Hu, X.; Zhang, H.; Wang, S.; Zhang, H.; You, C.; Zhang, C.Y.; Liang, H.; Chen, X.; Ba, Y. MiR-193a-3p is an important tumour suppressor in lung cancer and directly targets KRAS. Cell Physiol. Biochem. 2017, 44, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.T.; Borchert, G.M. Computational prediction of microRNA target genes, target prediction databases, and web resources. Methods Mol. Biol. 2017, 1617, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Liu, C.; Min, L.; Kuang, J.; Zhu, C.; Qiu, X.Y.; Zhu, L. Bioinformatic identification of miR-622 key target genes and experimental validation of the miR-622-RNF8 axis in breast cancer. Front. Oncol. 2019, 9, 1114. [Google Scholar] [CrossRef]
- Lukasik, A.; Wójcikowski, M.; Zielenkiewicz, P. Tools4miRs—One place to gather all the tools for miRNA analysis. Bioinformatics 2016, 32, 2722–2724. [Google Scholar] [CrossRef]
- Ritchie, W.; Rasko, J.E.J. Refining microRNA target predictions: Sorting the wheat from the chaff. Biochem. Biophys. Res. Commun. 2014, 445, 780–784. [Google Scholar] [CrossRef]
- Elton, T.S.; Yalowich, J.C. Experimental procedures to identify and validate specific mRNA targets of miRNAs. EXCLI J. 2015, 14, 758–790. [Google Scholar] [PubMed]
- Kuhn, D.E.; Martin, M.M.; Feldman, D.S.; Terry, A.V.J.; Nuovo, G.J.; Elton, T.S. Experimental validation of miRNA targets. Methods 2008, 44, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Castiglioni, I. Portrait of tissue-specific coexpression networks of noncoding RNAs (miRNA and lncRNA) and mRNAs in normal tissues. Comput. Math. Methods Med. 2019, 9029351. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zheng, J.; Chen, Z.; Liu, Y.; Dura, B.; Kwak, M.; Xavier-Ferrucio, J.; Lu, Y.-C.; Zhang, M.; Roden, C.; et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat. Commun. 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Sansom, S.E.; Nuovo, G.J.; Martin, M.M.; Kotha, S.R.; Parinandi, N.L.; Elton, T.S. miR-802 regulates human angiotensin II type 1 receptor expression in intestinal epithelial C2BBe1 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G632–G642. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J. In situ detection of microRNAs in paraffin embedded, formalin fixed tissues and the colocalization of their putative targets. Methods 2010, 52, 307–315. [Google Scholar] [CrossRef]
- Martinez-Sanchez, A.; Murphy, C.L. MicroRNA target identification-experimental approaches. Biology 2013, 2, 189–205. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Jiang, J.; Liu, Q.; Yang, L. A High-Throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 2004, 32, e43. [Google Scholar] [CrossRef]
- Ponchel, F.; Toomes, C.; Bransfield, K.; Leong, F.T.; Douglas, S.H.; Field, S.L.; Bell, S.M.; Combaret, V.; Puisieux, A.; Mighell, A.J.; et al. Real-time PCR based on SYBR-Green I fluorescence: An alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003, 3, 18. [Google Scholar] [CrossRef]
- Forero, D.A.; González-Giraldo, Y.; Castro-Vega, L.J.; Barreto, G.E. qPCR-based methods for expression analysis of miRNAs. Biotechniques 2019, 67, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, F.E. Experimental validation of microRNA targets using a luciferase reporter system. Methods Mol. Biol. 2011, 732, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zou, Q.; Song, H.; Song, F.; Wang, L.; Zhang, G.; Shen, X. A study of miRNAs targets prediction and experimental validation. Protein Cell 2010, 1, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Lieberman, J.; Lal, A. Desperately seeking microRNA targets. Nat. Struct Mol. Biol. 2010, 17, 1169–1174. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Carroll, A.P.; Goodall, G.J.; Liu, B. Understanding principles of miRNA target recognition and function through integrated biological and bioinformatics approaches. Wiley Interdiscip. Rev. RNA 2014, 5, 361–379. [Google Scholar] [CrossRef]
- Karbiener, M.; Glantschnig, C.; Scheideler, M. Hunting needle in the haystack: A guide to obtain biologically meaningful microRNA targets. Int. J. Mol. Sci. 2014, 15, 20266–20289. [Google Scholar] [CrossRef]
- Arvey, A.; Larsson, E.; Sander, C.; Leslie, C.S.; Marks, D.S. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Biol. 2010, 6, 363. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef]
- Khan, A.A.; Betel, D.; Miller, M.L.; Sander, C.; Leslie, C.S.; Marks, D.S. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 2009, 27, 549–555. [Google Scholar] [CrossRef]
- Kim, S.K.; Nam, J.W.; Rhee, J.K.; Lee, W.J.; Zhang, B.T. MiTarget: microRNA target-gene prediction using a support vector machine. BMC Bioinform. 2006, 7, 411. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Bracken, C.P.; Goodall, G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2014, 39, 6845–6853. [Google Scholar] [CrossRef] [PubMed]
- Tarang, S.; Weston, M.D. Macros in microRNA target identification. A comparative analysis of in silico, in vitro and in vivo approaches to microRNA target identification. RNA Biol. 2014, 11, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced applications of RNA sequencing and challenges. Bioinform. Biol. Insights 2015, 9, 29–46. [Google Scholar] [CrossRef]
- Beitzinger, M.; Peters, L.; Zhu, J.Y.; Kremmer, E.; Meister, G. Identification of human microRNA targets from isolated Argonaute protein complexes. RNA Biol. 2007, 4, 76–84. [Google Scholar] [CrossRef]
- Beitzinger, M.; Meister, G. Experimental identification of microRNA targets by immunoprecipitation of Argonaute protein complexes. Methods Mol. Biol. 2011, 732, 153–167. [Google Scholar] [CrossRef]
- Hock, J.; Weinmann, L.; Ender, C.; Rudel, S.; Kremmer, E.; Raabe, M.; Urlaub, H.; Meister, G. Proteomic and functional analysis of Argonaute-containing mRNAprotein complexes in human cells. EMBO Rep. 2007, 8, 1052–1060. [Google Scholar] [CrossRef]
- Weinmann, L.; Hock, J.; Ivacevic, T.; Ohrt, T.; Mutze, J.; Schwille, P.; Kremmer, E.; Benes, V.; Urlaub, H.; Meister, G. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell 2009, 136, 496–507. [Google Scholar] [CrossRef]
- Riley, K.J.; Yario, T.A.; Steitz, J.A. Association of Argonaute proteins and microRNAs can occur after cell lysis. RNA 2012, 18, 1581–1585. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.J.; Jungkamp, A.C.; Munschauer, M. Transcriptome wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Kishore, S.; Jaskiewicz, L.; Burger, L.; Hausser, J.; Khorshid, M.; Zavolan, M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 2011, 8, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Darnell, R.B. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011, 29, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent non-canonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions—Beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Si, M.-L.; Wu, H.; Mo, Y.-Y. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J. Biol. Chem. 2007, 282, 14328–14336. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool Name | SF | TS | EC | SA | Web | Ref. |
|---|---|---|---|---|---|---|
| miRanda | √ | √ | √ | http://www.microrna.org/microrna | [19] | |
| TargetScan | √ | √ | √ | √ | http://www.targetscan.org/vert_72 | [29] |
| RNAhybrid | √ | https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid | [17] | |||
| PITA | √ | √ | https://genie.weizmann.ac.il/pubs/mir07/mir07_prediction.html | [30] | ||
| microTAR | √ | √ | http://tiger.dbs.nus.edu.sg/microtar | [16] | ||
| PicTar | √ | √ | https://pictar.mdc-berlin.de | [27] | ||
| PACMIT | √ | √ | √ | https://paccmit.epfl.ch | [31] | |
| MIRZA-G | √ | √ | √ | √ | http://www.clipz.unibas.ch/index.php?r=tools/sub/mirza_g | [32] |
| RNA22 | √ | √ | https://cm.jefferson.edu/rna22/Interactive | [33] |
| Tool Name | Algorithm | Positive | Negative | Features | Ref. |
|---|---|---|---|---|---|
| MBSTar | Random Forest | MiRbase | Randomly generated | sequence, structure | [40] |
| NbmiRTar | Naïve Bayes | TarBase | Probability Randomization | sequence | [41] |
| TargetBoost | Genetic Programming | let-7, lin-4, miR-13a, bantam | Random string | sequence | [42] |
| DeepTarget | RNN | miRecords miRBase | Mocking in alignment | sequence | [43] |
| TargetMiner | SVM | miRecords | Randomly generated | seed | [44] |
| DeepMirTar | Autoencoder | miRecords | Mocking in alignment | sequence, structure, energy and other | [45] |
| DIANA-microT-CDS | microT-CDS algorithm | miRNA regulatory element in both the 3’-UTR and CDS | sequence, structure, energy and other | [46] | |
| miRanda-mirSVR | SVR (similar to SVM) | set from transfection experiments | sequence, structure, energy and other | [47] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riolo, G.; Cantara, S.; Marzocchi, C.; Ricci, C. miRNA Targets: From Prediction Tools to Experimental Validation. Methods Protoc. 2021, 4, 1. https://doi.org/10.3390/mps4010001
Riolo G, Cantara S, Marzocchi C, Ricci C. miRNA Targets: From Prediction Tools to Experimental Validation. Methods and Protocols. 2021; 4(1):1. https://doi.org/10.3390/mps4010001
Chicago/Turabian StyleRiolo, Giulia, Silvia Cantara, Carlotta Marzocchi, and Claudia Ricci. 2021. "miRNA Targets: From Prediction Tools to Experimental Validation" Methods and Protocols 4, no. 1: 1. https://doi.org/10.3390/mps4010001
APA StyleRiolo, G., Cantara, S., Marzocchi, C., & Ricci, C. (2021). miRNA Targets: From Prediction Tools to Experimental Validation. Methods and Protocols, 4(1), 1. https://doi.org/10.3390/mps4010001