A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS

 ,
,
Abstract
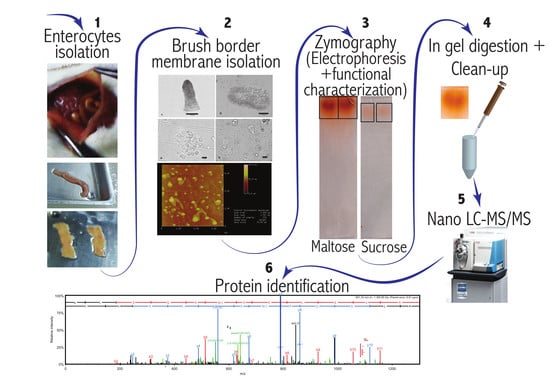
1. Introduction
2. Material and Methods
2.1. Brush Border Membrane Preparation and Zymography
2.2. Enzymatic “In Gel” Digestion
2.3. Nano LC-MS/MS
3. Data Analysis and Protein Identification
4. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morales, R.L.C.; Zazueta-Novoa, V.; Leal-Morales, C.A.; Martínez, A.F.; Noyola, P.P.; Zazueta-Sandoval, R. Polyacrylamide gel electrophoresis an important tool for the detection and analysis of enzymatic activities by electrophoretic zymograms. In Gel Electrophoresis—Advances Techniques; Magdelin, S., Ed.; InTech: Rijeka, Croatia, 2012; p. 500. [Google Scholar]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase–Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Jones, K.; Sim, L.; Mohan, S.; Kumarasamy, J.; Liu, H.; Avery, S.; Naim, H.Y.; Quezada-Calvillo, R.; Nichols, B.L.; Mario Pinto, B.; et al. Mapping the intestinal alpha-glucogenic enzyme specificities of starch digesting maltase-glucoamylase and sucrase-isomaltase. Bioorg. Med. Chem. 2011, 19, 3929–3934. [Google Scholar] [CrossRef]
- Nichols, B.L.; Eldering, J.; Avery, S.; Hahn, D.; Quaroni, A.; Sterchi, E. Human small intestinal maltase-glucoamylase cDNA cloning: Homology to sucrase-isomaltase. J. Biol. Chem. 1998, 273, 3076–3081. [Google Scholar] [CrossRef]
- Nichols, B.L.; Avery, S.; Sen, P.; Swallow, D.M.; Hahn, D.; Sterchi, E. The maltase-glucoamylase gene: common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc. Natl. Acad. Sci. USA 2003, 100, 1432–1437. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef]
- Ernst, H.A.; Lo Leggio, L.; Willemoës, M.; Leonard, G.; Blum, P.; Larsen, S. Structure of the Sulfolobus solfataricus α-Glucosidase: Implications for domain conservation and substrate recognition in GH31. J. Mol. Biol. 2006, 358, 1106–1124. [Google Scholar]
- Chaudet, M.M.; Amiri, M.; Marth, N.; Naim, H.Y.; Rose, D.R. Phylogenetic analysis reveals key residues in substrate hydrolysis in the isomaltase domain of sucrase-isomaltase and its role in starch digestion. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1410–1416. [Google Scholar] [CrossRef]
- Rose, D.R.; Chaudet, M.M.; Jones, K. Structural studies of the intestinal α-glucosidases, maltase-glucoamylase and sucrase-isomaltase. J. Pediatr. Gastroenterol. Nutr. 2018, 66, S11–S13. [Google Scholar] [CrossRef]
- Mac Donal, O.; Chediack, J.G.; Caviedes-Vidal, E. Isolation of epithelial cells, villi and crypts from small intestine of pigeons (Columba livia). Biocell 2008, 32, 219–227. [Google Scholar]
- McConnell, R.E.; Benesh, A.E.; Mao, S.; Tabb, D.L.; Tyska, M.J. Proteomic analysis of the enterocyte brush border. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G914–G926. [Google Scholar] [CrossRef]
- Simpson, R.J. Staining proteins in gels with coomassie blue. CSH Protoc. 2007, 2007, pdb.prot4719. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Wang, G.; de Jong, R.N.; van den Bremer, E.T.J.; Parren, P.W.H.I.; Heck, A.J.R. Enhancing accuracy in molecular weight determination of highly heterogeneously glycosylated proteins by native tandem mass spectrometry. Anal. Chem. 2017, 89, 4793–4797. [Google Scholar] [CrossRef]
- Gericke, B.; Amiri, M.; Naim, H.Y. The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol. Cell. Pediatr. 2016, 3, 2. [Google Scholar] [CrossRef]
- Biviano, A.B.; Del Río, C.M.; Phillips, D.L. Ontogenesis of intestine morphology and intestinal disaccharidases in chickens (Gallus gallus) fed contrasting purified diets. J. Comp. Physiol. B 1993, 163, 508–518. [Google Scholar]
- Lee, B.H.; Rose, D.R.; Lin, A.H.M.; Quezada-Calvillo, R.; Nichols, B.L.; Hamaker, B.R. Contribution of the individual small intestinal α-glucosidases to digestion of unusual α-linked glycemic disaccharides. J. Agric. Food Chem. 2016, 64, 6487–6494. [Google Scholar] [CrossRef]
- Semenza, G.; Auricchio, S.; Mantei, N. Small-Intestinal Disaccharidases. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M., Mitchell, G., Eds.; The McGraw-Hill Companies, Inc.: New York, NY, USA, 2014. [Google Scholar]
- Brun, A.; Mendez-Aranda, D.; Magallanes, M.E.; Karasov, W.H.; Martinez Del Rio, C.; Baldwin, M.; Caviedes-Vidal, E. Evolution of intestinal α-glucosidases in vertebrates: Genomic and proteomic data upend previous hypotheses. Integr. Comp. Biol. 2019, 59, E25. [Google Scholar]


{kind=link}
{kind=link}
| Electrophoresis | ||
|---|---|---|
| Gel | I | II |
| Constituents 1 | 4–12% mini polyacrylamide gel 1 | |
| Temperature of the run | room temperature | |
| Total protein loaded per well | 10 µg | |
| Running buffer | Tris-glycine pH 8.3 | |
| Electric field applied | 100 V constant and ~350 mA | |
| Run time | 3 h | |
| Hydrolytic activity assay | ||
| Substrate solution and concentration | Maltose 56 mM | Sucrose 56 mM |
| Incubation time | 1 h | 1 h |
| Incubation temperature | 37 °C | |
| Assay reagent 2 Incubation time (reaction temperature) | 1 h (37 °C) | 5 h (37 °C) |
| Rinse the gel with deionized water | 3 × | |
| Gel storage for further analysis | 4 °C | |
| Enzyme | Uniprot Accesion Number | Molecular Mass (kDa) | Maltose Substrate | Sucrose Substrate | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mouse 1 | Mouse 2 | Mouse 1 | Mouse 2 | |||||||
| Quantitative Value (Normalized Total Spectra) | Coverage % | Quantitative Value (Normalized Total Spectra) | Coverage % | Quantitative Value (Normalized Total Spectra) | Coverage % | Quantitative Value (Normalized Total Spectra) | Coverage % | |||
| SI (sucrase-isomaltase) | F8VQM5 | 209 | 104 | 23 | 42 | 3 | 100 | 35 | 81 | 6 |
| MGAM (maltase-glucoamylase) | B5THE2 | 209 | 56 | 15 | 94 | 6 | 83 | 31 | 38 | 4 |
| ANPEP (aminopeptidase-N) | P97449 | 110 | 25 | 11 | 42 | 2 | 49 | 39 | 49 | 5 |
| ENPEP (Glutamyl Aminopeptidase) | P16406 | 108 | 9 | 8 | - | - | 12 | 21 | - | - |
| DPP4 (Dipeptidyl peptidase 4) | P28843 | 87 | 3 | 4 | - | - | 7 | 16 | - | - |
| MEP1B (Meprin A Subunit Beta) | Q61847 | 80 | 3 | 10 | - | - | 5 | 12 | - | - |
| All other non-hydrolase proteins (details in Supplementary Table S1) | 194 | 147 | 210 | 211 | ||||||
| Hydrolases as % of total proteins | 51% | 55% | 55% | 44% | ||||||
| α-glucosidases as % of total hydrolases | 80% | 76% | 71% | 71% | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brun, A.; Magallanes, M.E.; Martínez del Rio, C.; Barrett-Wilt, G.A.; Karasov, W.H.; Caviedes-Vidal, E. A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS. Methods Protoc. 2020, 3, 15. https://doi.org/10.3390/mps3010015
Brun A, Magallanes ME, Martínez del Rio C, Barrett-Wilt GA, Karasov WH, Caviedes-Vidal E. A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS. Methods and Protocols. 2020; 3(1):15. https://doi.org/10.3390/mps3010015
Chicago/Turabian StyleBrun, Antonio, Melisa E. Magallanes, Carlos Martínez del Rio, Gregory A. Barrett-Wilt, William H. Karasov, and Enrique Caviedes-Vidal. 2020. "A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS" Methods and Protocols 3, no. 1: 15. https://doi.org/10.3390/mps3010015
APA StyleBrun, A., Magallanes, M. E., Martínez del Rio, C., Barrett-Wilt, G. A., Karasov, W. H., & Caviedes-Vidal, E. (2020). A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS. Methods and Protocols, 3(1), 15. https://doi.org/10.3390/mps3010015