Reverse Genetic Systems for Pseudomonas aeruginosa Leviphages


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
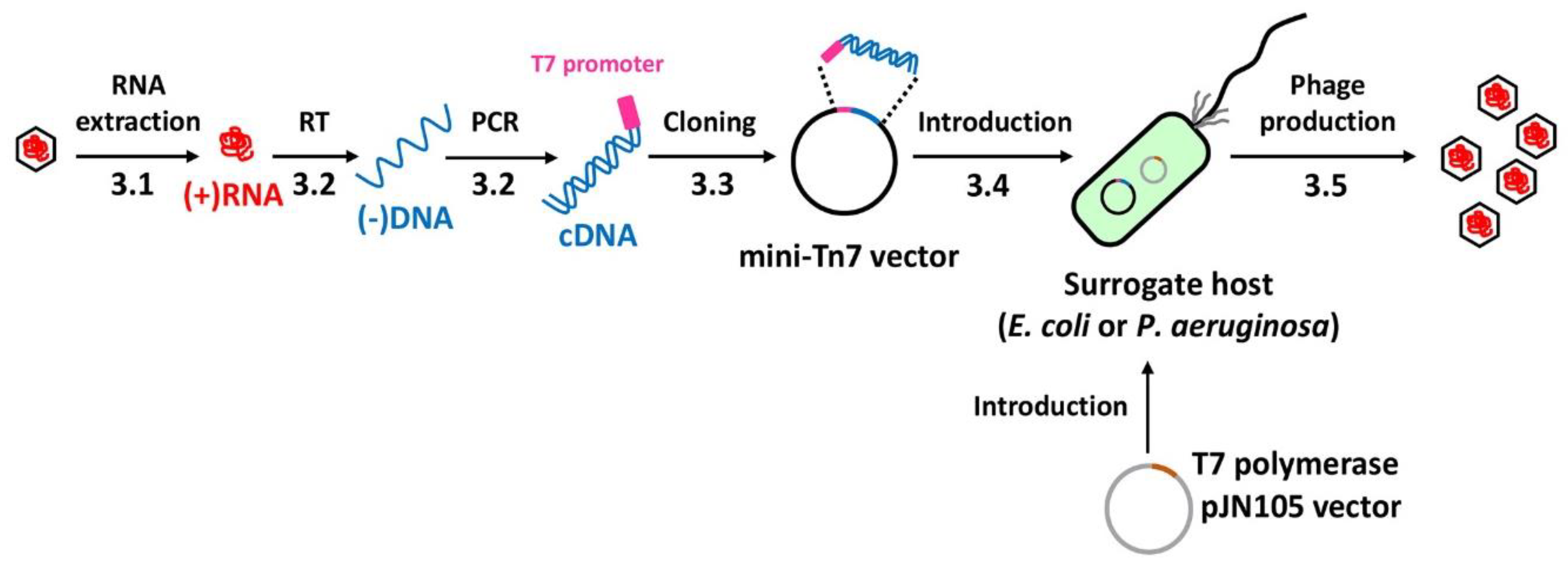
:1. Introduction
2. Experimental Design
2.1. Reagents
- RNase free water (Qiagen, Hilden, Germany; Cat. no.: 129112)
- TRIZOL (Ambion, Austion, TX, USA; Cat. no.: 15596026)
- Sodium chloride (DAEJUNG, Siheung, Korea; Cat. no.: 7548-4400)
- Potassium chloride (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: P3911-1KG)
- Calcium chloride dihydrate (Sigma-Aldrich; Cat. no.: C3306-500G)
- Magnesium chloride hexahydrate (Sigma-Aldrich; Cat. no.: M9272-500G)
- Magnesium sulfate heptahydrate (Sigma-Aldrich; Cat. no.: M1880-500G)
- Tris-HCl, pH 7.5 (Sigma-Aldrich; Cat. no.: T2663-1L)
- Ethanol (EMSURE, Darmstadt, Germany; Cat. no.: 1.00983.1011)
- Chloroform (Junsei, Tokyo, Japan; Cat. no.: 28560S0350)
- Sucrose (Junsei; Cat. no.: 31365S0301)
- RNase-free DNase I set (Qiagen; Cat. no.: 79254)
- RNeasy MinElute clean-up kit (Qiagen; Cat. no.: 74204)
- Exprep Plasmid SV mini kit (Geneall, Seoul, Korea; Cat. no.: 101-102)
- Superiorscript III Reverse Transcriptase (Enzynomics, Daejeon, Korea; Cat. no.: RT006M)
- 5× First-Strand buffer (Enzynomics; Cat. no.: RT006M)
- dNTP mixture (10 mM) (Enzynomics; Cat. no.: RT006M)
- 0.1 M DTT (Enzynomics; Cat. no.: RT006M)
- RNase inhibitor (Enzynomics; Cat. no.: RT006M)
- Phusion, High Fidelity DNA polymerase (Thermo Fisher, Vilnius, Lithuania; Cat. no.: F530L)
- 5× High Fidelity buffer (Thermo Fisher; Cat. no.: F530L)
- dNTP mixture (2.5 mM) (Takara bio, Shiga, Japan; Cat. no.: 4030)
- Dimethyl sulfoxide (DAEJUNG; Cat. no.: 3047-4400)
- SpeI (Enzynomics; Cat. no.: R011S)
- HindIII (New England Biolabs, Ipswich, MA, USA; Cat. no.: R0104S)
- 2.1 10× buffer (New England Biolabs; Cat. no.: B7202S)
- T4 ligase (New England Biolabs; Cat. no.: M0202M)
- T4 ligase 10× buffer (New England Biolabs; Cat. no.: B0202S)
- Expin PCR SV (Geneall; Cat. no.: 103-102)
- Expin Gel SV (Geneall; Cat. no.: 102-102)
- Terminal deoxynucleotidyl transferase (Thermo Fisher; Cat. no.: EP0161)
- 5× A-tailing buffer (Thermo Fisher; Cat. no.: EP0161)
- dATP (2 mM) (Cosmo genetech, Seoul, Korea; Cat. no.: NT050)
- Phage buffer (0.1 M NaCl, 50 mM Tris-HCl (pH7.5), 0.01 M MgSO4·7H2O)
- 5× KCM buffer (0.5 M KCl, 0.15 M CaCl2, 0.25 M MgCl2)
- Tryptone (NEOGEN, Lansing, MI, USA; Cat. no.: 7351B)
- Yeast extract (NEOGEN; Cat. no.: 7184A)
- Agar (NEOGEN; Cat. no.: 7188A)
- Cetrimide agar (Becton Dickinson, Le Pont de Claix, France; Cat. no.: 285420)
- Agarose (elbio, Seongnam, Korea; Cat. no.: CA007)
- Gentamicin sulfate (MBcell, Seoul, Korea; Cat. no.: MB-G4582)
- Carbenicillin disodium (Duchefa Biochemie, Haarlem, Netherlands; Cat. no.: C0109)
- LB broth: 1% tryptone, 0.5% yeast extract, 1% NaCl
- LB plate: 1% tryptone, 0.5% yeast extract, 1% NaCl, 2% Bacto-agar
- Cetrimide agar (CA) plate (for Pseudomonas isolation): 4.53% cetrimide agar (Difco), 1% glycerol
- Distilled water
- 70% Ethanol
2.2. Equipment
- Centrifuge (Eppendorf, Hamburg, Germany; Cat. no.: 5415R)
- Thermal cycler (Bio-RAD, Hercules, CA, USA; Cat. no.: C1000)
- Incubator (Hanbaek Scientific, Bucheon, Korea)
- Shaking Incubator (Hanbaek Scientific)
- Heating block (WEALTech, New Taipei City, Taiwan; Cat. no.: HB-1)
- Eppendorf tubes (Sarstedt, Nümbrecht, Germany)
- Centrifuge (Eppendorf)
- Spectrophotometer (ND-1000, Eppendorf)
- MicroPulser (BIO-RAD; Cat. No.: 411BR7556)
3. Procedure
3.1. RNA Extraction (Time for completion: 1 Day)
- Adjust the phage lysate containing ~1011 pfu of PP7 to 100 μL using phage buffer in a 1.5 mL Eppendorf tube.
- 2.
- Add 1 mL TRIZOL reagent and mix by pipetting.
- 3.
- Incubate the mixture at room temperature for 5 min.
- 4.
- Add 200 μL of chloroform, vortex vigorously for 15 s.
- 5.
- Incubate the mixture at room temperature for 3 min.
- 6.
- Centrifuge the mixture at 4 °C for 15 min at 12,000× g
- 7.
- Transfer 300 μL of the aqueous phase to a fresh 1.5 mL Eppendorf tube.
- 8.
- Add 750 μL of 100% ethanol to precipitate the RNA
- 9.
- Incubate the mixture at −20 °C for for 1 h
- 10.
- Centrifuge the mixture at 4 °C for 10 min at 12,000× g and discard the supernatant
- 11.
- Wash the RNA pellet twice with 1 mL of 70% ethanol
- 12.
- Centrifuge the mixture at 4 °C for 5 min at 7500× g and discard the supernatant
- 13.
- Remove the supernatant and dry the RNA pellet (air-dry) for 5 min.
- 14.
- Dissolve the RNA with RNase-free water (20 μL) and incubate for 10 min at 55–60 °C in a heating block.
3.2. cDNA Synthesis (Time for completion: 1 Day)
- Adjust the RNA solution (from 3.1) to a volume of 85 μL with RNase-free water in a 1.5 mL Eppendorf tube.
- Add 10 μL RDD buffer and 5 μL DNase I from RNase-free DNase set (Qiagen) to the RNA sample (100 μL in total).
- 3.
- Incubate the RNA sample at room temperature for 20 min.
- 4.
- Clean up the RNA sample with RNeasy MinElute Cleanup kit (Qiagen), according to the manufacturer’s instruction.
- 5.
- Transfer the DNase-treated RNA sample (less than 5 μg) to an 0.2 mL thin-wall PCR tube and incubate in a thermal cycler at 65 °C for 5 min.
- 6.
- Set up the RT (reverse transcription) mixture in 0.2 mL thin-wall PCR tube as follows (20 μL in total):
Template RNA diluted in RNase-free water 10.5 μL 5× First-Strand buffer 4 μL dNTP mixture (10 mM) 1 μL 0.1M DTT 2 μL RNase inhibitor (40 units/μL) 0.5 μL RT-Primer (PP7-3588R-H) (10 μM) 1 μL SuperiorScript III Reverse Transcriptase (200 units/μL) 1 μL - 7.
- Incubate the mixture in a thermal cycler at the following cycle setting: 50 °C for 45 min → 70 °C for 15 min → 4 °C
- 8.
- Set up the PCR mixture in 0.2 mL thin-wall PCR tube as follows (50 μL in total):
Template cDNA (aliquot from step 7) diluted in water 28 μL 5× High Fidelity buffer 10 μL dNTP mixture (2.5 mM) 4 μL DMSO 2.5 μL Forward primer (T7PP7-F-S) (10 μM) 2.5 μL Reverse primer: RT-Primer (PP7-3588R-H) (10 μM) 2.5 μL Phusion, High Fidelity DNA Polymerase (2 units/μL) 0.5 μL - 9.
- Incubate the mixture in a thermal cycler at the following cycle setting:
- Hot start: 98 °C for 30 s
- Amplification (30 cycles): 98 °C for 10 s → 60 °C for 30 s → 72 °C for 2 min
- Final extension: 72 °C for 5 min → 4 °C
- 10.
- Perform 0.8% agarose gel electrophoresis using the PCR sample.
- 11.
- Perform cDNA gel extraction using the Expin Gel SV kit.
3.3. cDNA Cloning (Time for completion: 2 Days)
- Set up the digestion mixture in 1.5 mL Eppendorf tube as follows (100 μL in total):
Insert cDNA (aliquot from 3.2) or vector DNA diluted in water 85 μL 10× Digestion buffer (2.1 NEB) 10 μL Restriction enzyme, SpeI (10 units/μL Enzynomics) 2.5 μL Restriction enzyme, HindIII (20 units/μL NEB) 2.5 μL - Incubate the digestion mixture at 37 °C for 3 h.
- Clean up the digested DNA samples (either insert or vector) using the Expin PCR SV kit according to the manufacturer’s instruction.
- Set up the ligation mixture in a 1.5-mL Eppendorf tube as follows (10 μL in total):
Insert (60–90 ng) and vector (20–30 ng) DNA diluted in water 8 μL 10× T4 DNA ligase buffer 1 μL T4 DNA ligase 1 μL
- 5.
- Incubate the ligation mixture at room temperature for 1 h.
- 6.
- Add 20 μL 5× KCM buffer and 70 μL TDW to the ligation mixture.
- 7.
- Add 100 μL competent cell (E. coli HB101) to the ligation mixture and gently mix.
- 8.
- Place the mixture on ice for 10 min.
- 9.
- Place the mixture at the heat block at 42 °C for 1.5 min
- 10.
- Place the mixture on ice for 2 min.
- 11.
- Add 1 mL LB to the mixture and incubate at 30 °C for 1 h in a shaking incubator.
- 12.
- Centrifuge the mixture at 8000× g for 5 min and discard supernatant.
- 13.
- Resuspend the cell pellet with 200 μL LB broth and spread on LB agar plate containing gentamicin (25 μg/mL).
- 14.
- Incubate the LB plate at 30 °C for 24–36 h.
- 15.
- Pick the single colonies for isolation of the stable transformants, grow them in LB broth, and make the glycerol (20%) stock for storage.
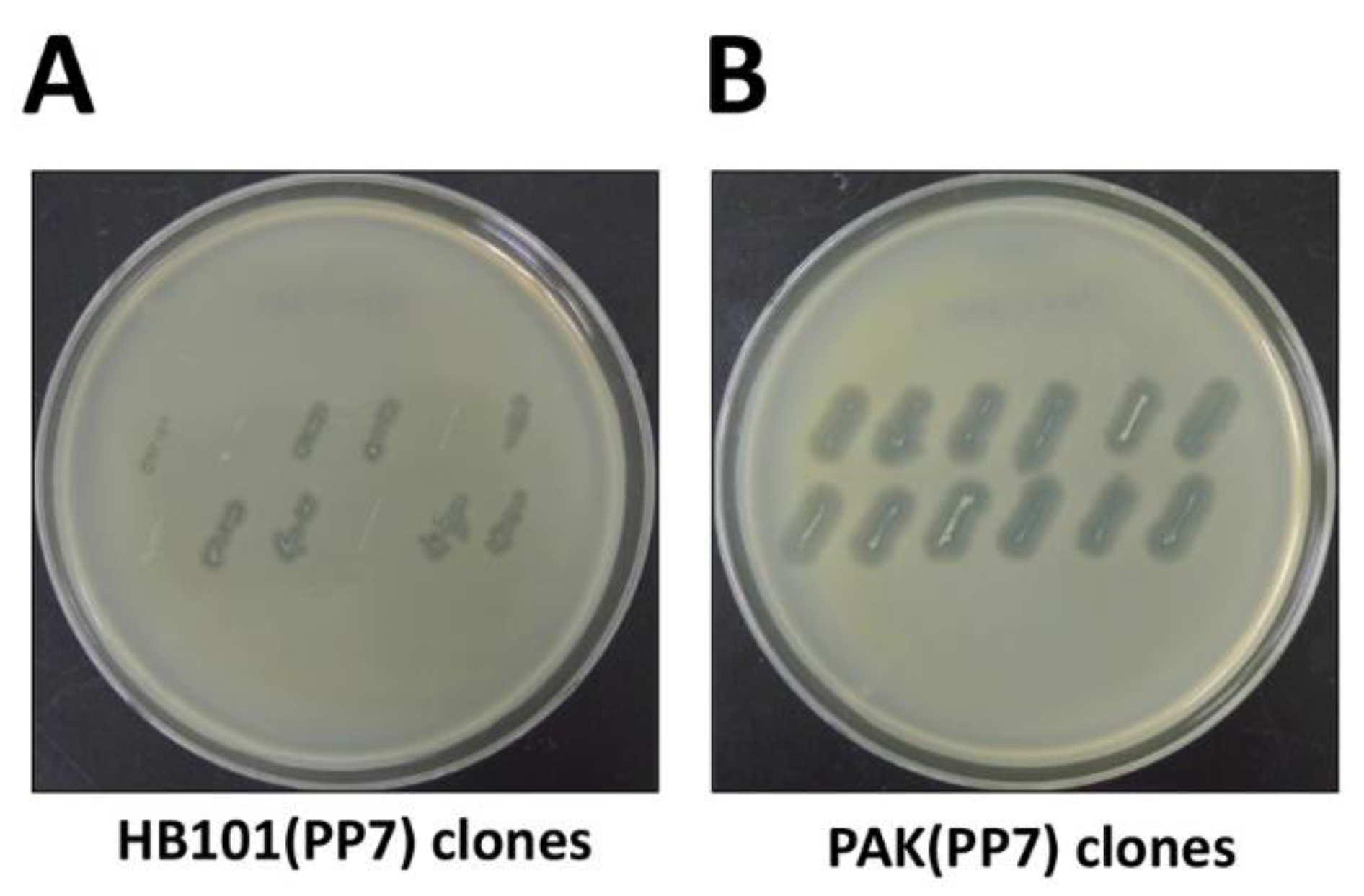
3.4. cDNA Introduction (Time for completion: 6~7 Days)
- Streak frozen glycerol stocks of the parental bacteria onto LB plates or CA plates (only for P. aeruginosa) and incubate the plates overnight at 37 °C (except for the HB101(PP7) that should be grown at 30 °C) to obtain fresh colonies.
- 2.
- Inoculate culture tubes containing 3 mL LB broth with fresh single colonies and incubate the tubes overnight in shaking incubator.
- 3.
- Inoculate culture tubes containing 3 mL LB broth by 1/100 diluting the seed culture and incubate the tubes until the OD600 reaches 3.0.
- 4.
- Centrifuge 1 mL of each culture aliquot at 8000× g for 5 min and discard the supernatant.
- 5.
- Wash the cell pellets with 1 mL of fresh LB broth.
- 6.
- Resuspend the cell pellets with 1 mL of LB broth and collect 200 μL of each cell suspension into a 1.5 mL Eppendorf tube (800 μL in total).
- 7.
- Centrifuge the cell mixture at 8000× g for 5 min and resuspend with 20 μL of LB broth.
- 8.
- Spread the resuspended cell mixture onto LB plate and incubate at 30 °C for overnight.
- 9.
- Scrape the cells from the LB plate with a scraper and resuspend in 200 μL of LB broth.
- 10.
- Spread the cells onto the CA plate containing gentamicin (50 μg/mL).
- 11.
- Incubate the CA plate at 30 °C for 24–36 h.
- 12.
- Pick the single exconjugant colonies for isolation of the stable exconjugants and make the glycerol (20%) stock for storage.
- 13.
- Inoculate culture tubes containing 3 mL broth with fresh colonies of the stable exconjugants and incubate the tubes overnight in a shaking incubator.
- 14.
- Inoculate culture tubes containing 3 mL LB broth by 1/100 diluting the seed culture and incubate the tubes until the OD600 reaches 0.8.
- 15.
- Centrifuge 3 mL of culture aliquot at 8000× g for 10 min and discard the supernatant.
- 16.
- Wash the cell pellets with 1 mL of 10% sucrose solution.
- 17.
- Resuspend the cell pellets with 200 μL of 10% sucrose solution in a 1.5 mL Eppendorf tube.
- 18.
- Electroporate the cell suspension with 50 ng of pFLP2 DNA using MicroPulser.
- 19.
- Incubate the cells at 30 °C for 1 h in shaking incubator.
- 20.
- Spread the cells onto the LB plate containing carbenicillin (200 μg/mL).
- 21.
- Incubate LB plate at 30 °C for 24–36 h
- 22.
- Check single colonies for antibiotic susceptibility by replicating each onto LB with gentamicin (50 μg/mL) and LB with carbenicillin (200 μg/mL).
- 23.
- Incubate 30 °C for overnight or until the colonies appear.
- 24.
- Streak carbenicillin-resistant but gentamicin-sensitive colonies onto an LB plate containing 5% sucrose.
- 25.
- Incubate overnight at 30 °C or until sucrose-resistant colonies appear.
- 26.
- Isolate the appropriate (i.e., sucrose-resistant, carbenicillin-sensitive, gentamicin-sensitive) stable single colonies and make glycerol (20%) stocks for storage.
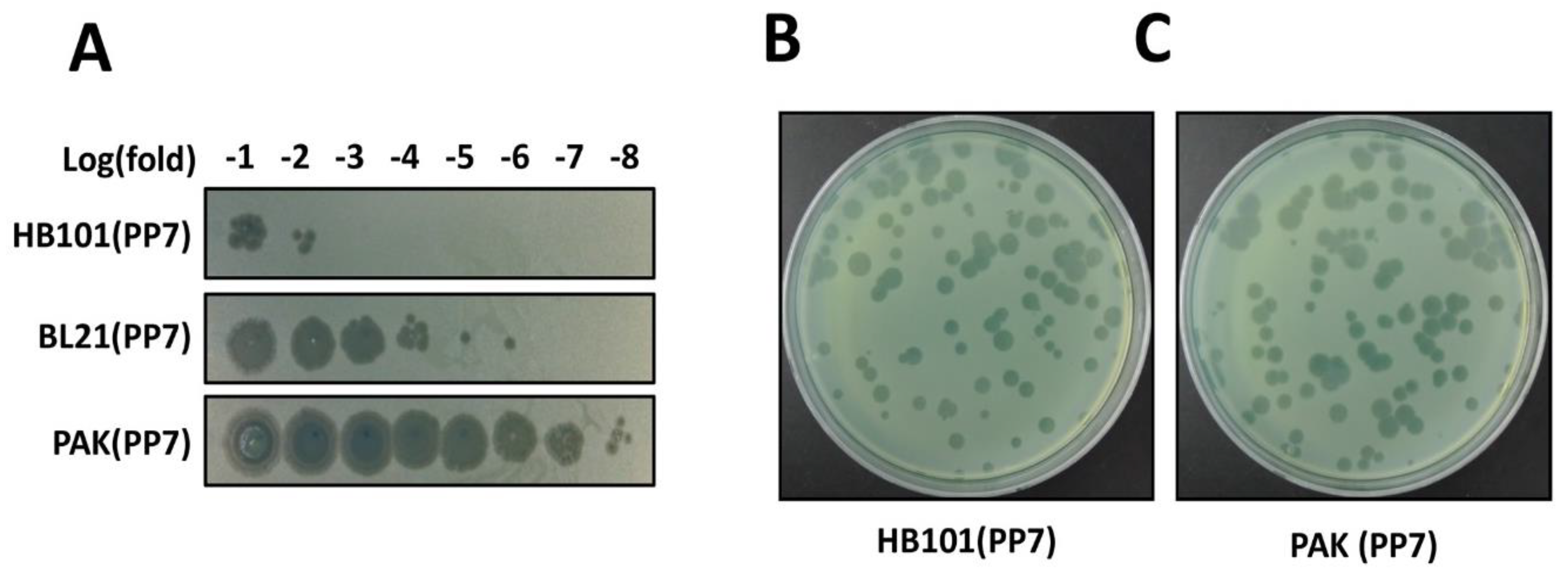
3.5. Phage Production (Time for completion: 3 Days)
- Streak frozen glycerol stocks of the PP7-producing bacteria onto LB plates and incubate the plates overnight at 30 °C to obtain fresh colonies. Likewise, the PP7-suceptible P. aeruginosa PAO1 is prepared in parallel except for incubation at 37 °C.
- 2.
- Inoculate culture tubes containing 3 mL LB broth with fresh single colonies and incubate the tubes overnight in a shaking incubator.
- 3.
- Transfer the PP7-producer culture to a 1.5 mL Eppendorf tube and add 0.1% CHCl3.
- 4.
- Centrifuge the cultures at 12,000× g for 10 min and obtain the culture supernatant for the phage sample.
- 5.
- For phage titration, the phage sample is serially diluted in phage buffer.
- 6.
- Add the overnight-grown PAO1 cells (50 μL) to the 3 mL top agar and vortex briefly.
- 7.
- Pour the top agar onto the pre-equilibrated LB plate and store 30 min at the clean bench.
- 8.
- Spot the serially diluted phage samples on the LB plate.
- 9.
- Incubate the LB plate at 37 °C for overnight (12–18 h).
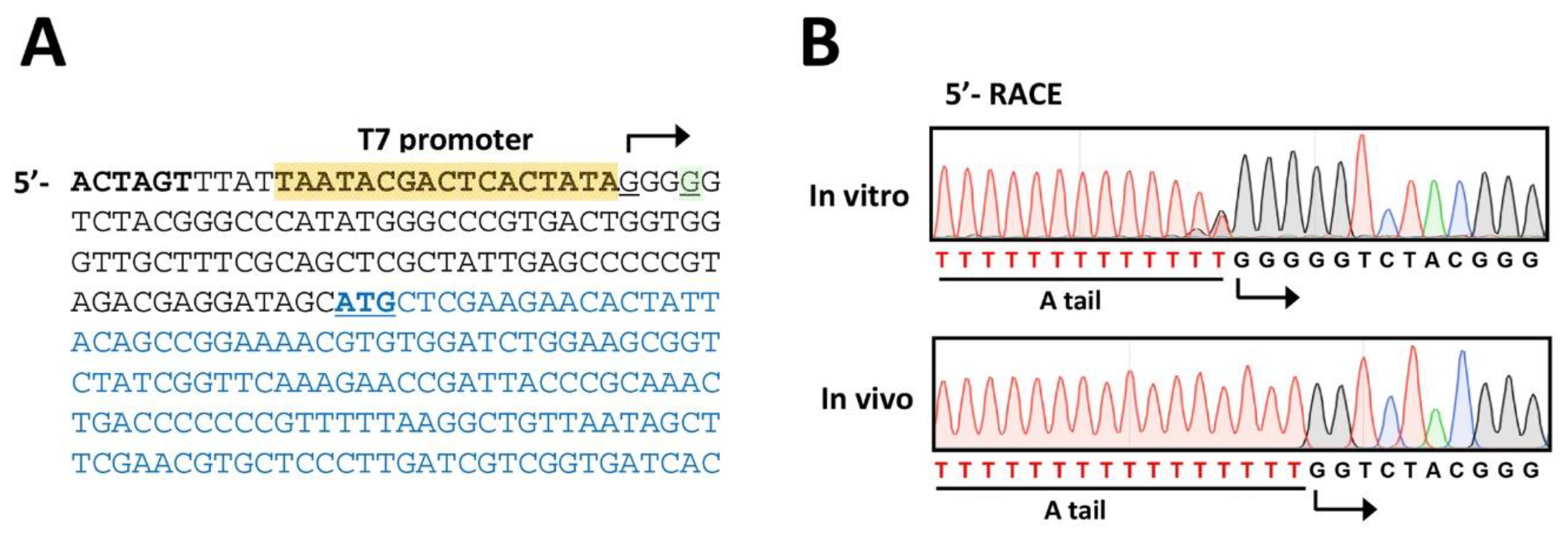
3.6. Phage Verification (Time for completion: 1~2 Days)
- Perform the steps of 3.1 RNA Extraction using the phage samples obtained from 3.5. Phage Production.
- 2.
- Perform the steps 1 to 5 of 3.2 cDNA Synthesis.
- 3.
- Set up the RT mixture in 0.2 mL thin-wall PCR tube as follows (20 μL in total):
Template RNA diluted in RNase-free water 10.5 μL 5× First-Strand buffer 4 μL dNTP mixture (10 mM) 1 μL 0.1M DTT 2 μL RNase inhibitor (40 units/μL) 0.5 μL RT-Primer (PP7-340R) (10 μM) 1 μL SuperiorScript III Reverse Transcriptase (200 units/μL) 1 μL - 4.
- Incubate the mixture in a thermal cycler at the following cycle setting: 50 °C for 45 min → 70 °C for 15 min → 4 °C
- 5.
- Perform cDNA extraction using the Expin PCR SV kit.
- 6.
- Set up the A-tailing mixture in 0.2 mL thin-wall PCR tube as follows (20 μL in total):
Template cDNA (aliquot from step 5) (200 ng) in water 13.5 μL 5× A-tailing buffer 4 μL dATP (2 mM) 1 μL Terminal deoxynucleotidyl transferase (20 units/μL) 1.5 μL - 7.
- Incubate the mixture at 37 °C for 30 min and then at 70 °C for 10 min.
- 8.
- Set up the 5′-RACE mixture in 0.2 mL thin-wall PCR tube as follows (50 μL in total):
Template cDNA (aliquot from step 7) diluted in water 28 μL 5× High Fidelity buffer 10 μL dNTP mixture (2.5 mM) 4 μL DMSO 2.5 μL Forward primer (PP7-319R) (10 μM) 2.5 μL Reverse primer (dT adapter) (10 μM) 2.5 μL Phusion, High Fidelity DNA Polymerase (2 units/μL) 0.5 μL - 9.
- Incubate the mixture in a thermal cycler at the following cycle setting:
- Hot start: 98 °C for 30 s
- Amplification (30 cycles): 98 °C for 10 s → 60 °C for 30 s → 72 °C for 2 min
- Final extension: 72 °C for 5 min → 4 °C
- 10.
- Perform 0.8% agarose gel electrophoresis using the 5′-RACE product.
- 11.
- Perform DNA extraction using the Expin Gel SV kit for sequence verification.
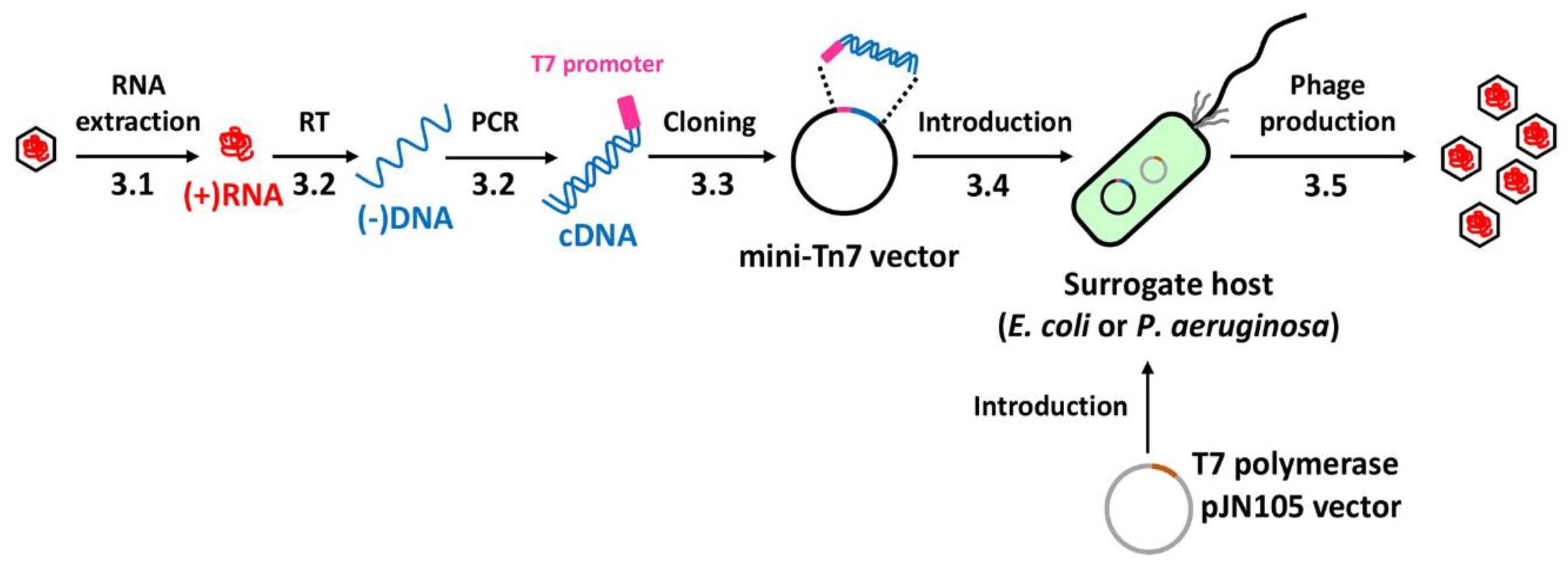
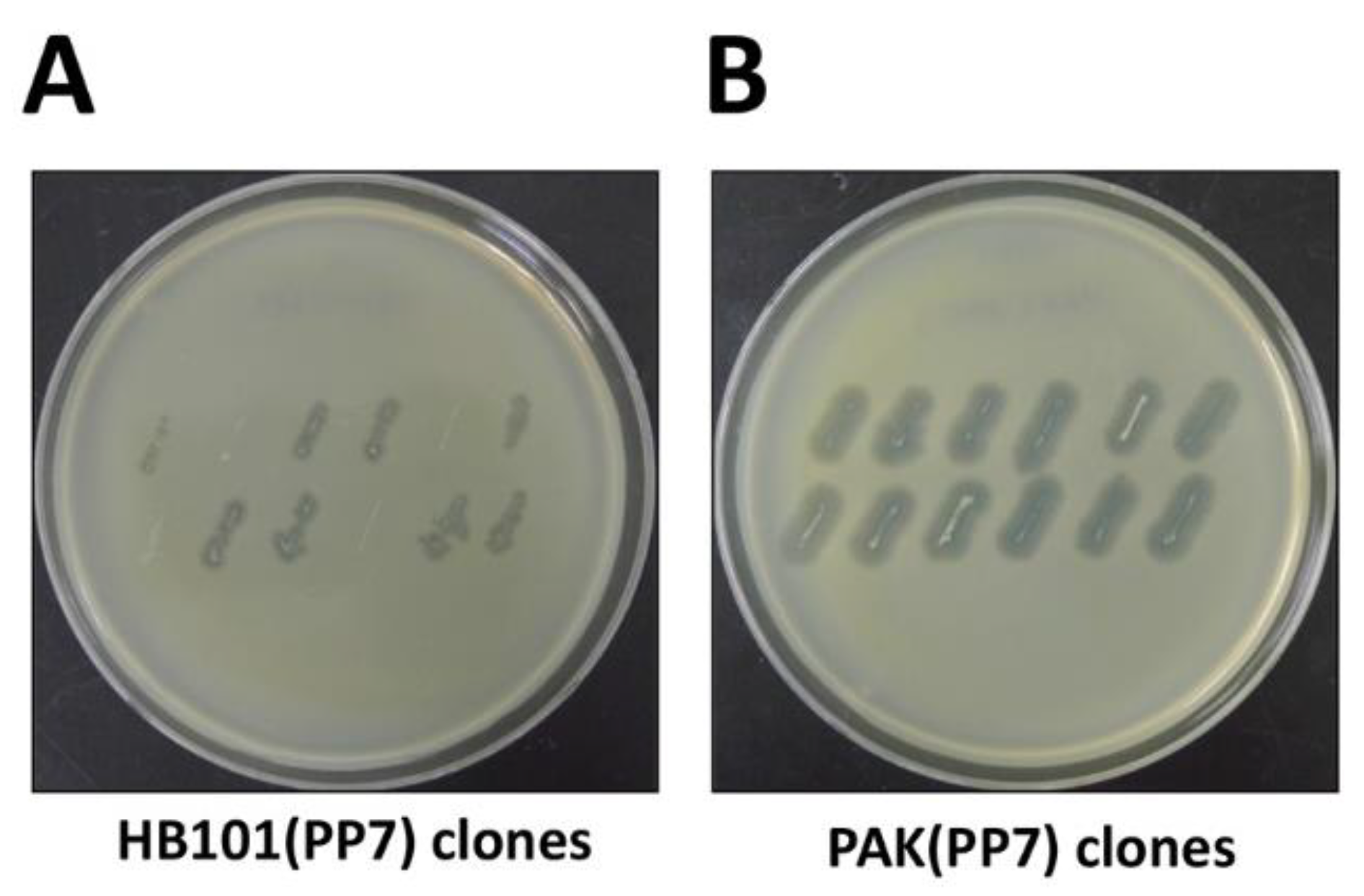
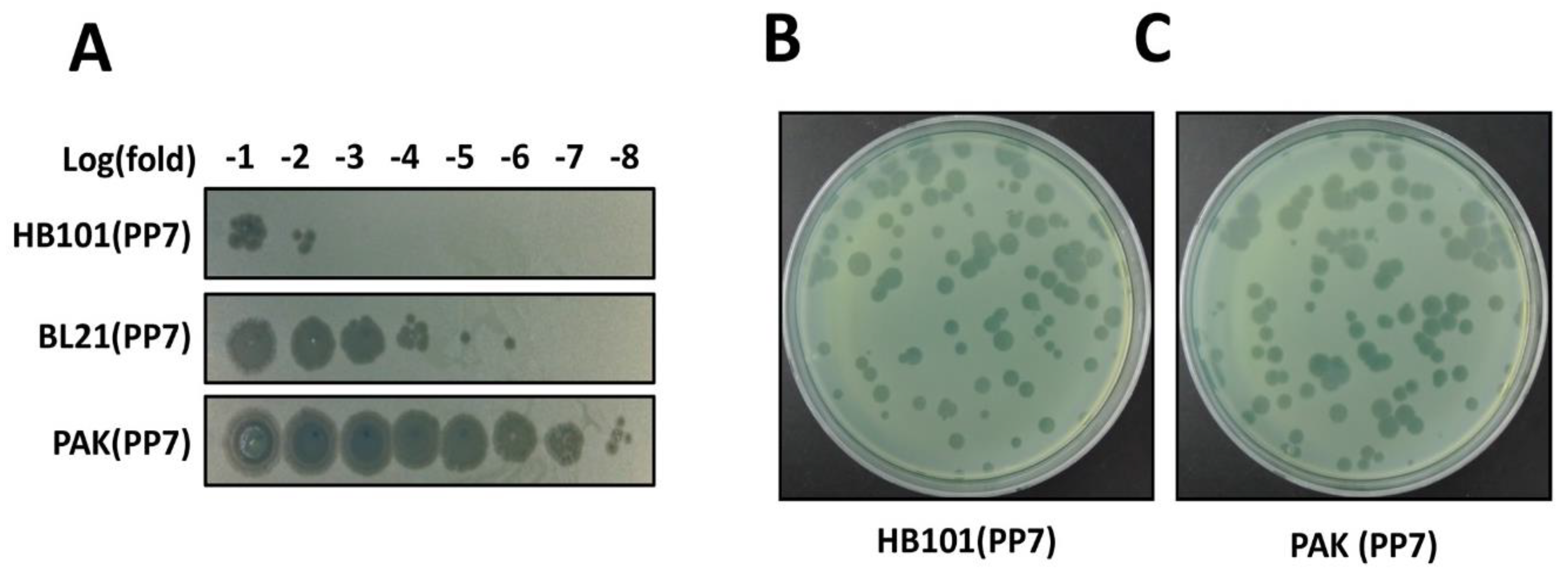
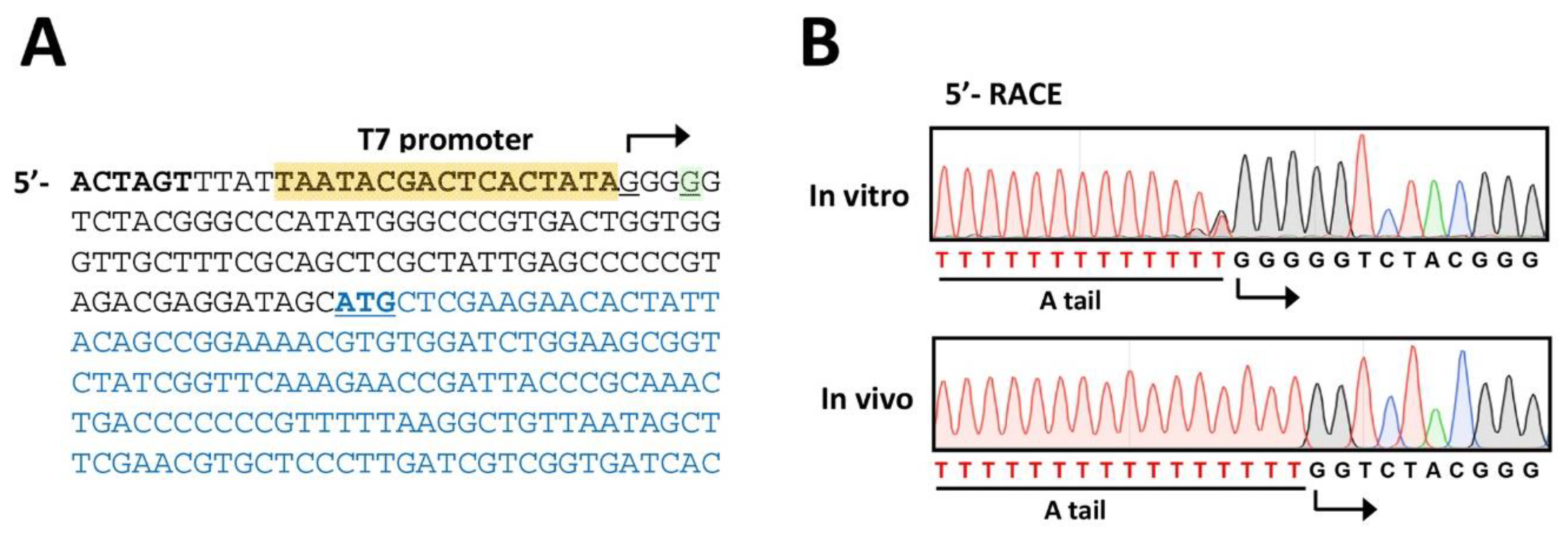
4. Expected Results
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Srinivasiah, S.; Bhavsar, J.; Thapar, K.; Liles, M.; Schoenfeld, T.; Wommack, K.E. Phages across the biosphere: Contrasts of viruses in soil and aquatic environments. Res. Microbiol. 2008, 159, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Piers, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically engineered phages: A review of advances over the last decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Lemire, S.; Yehl, K.M.; Lu, T.K. Phage-based applications in synthetic biology. Annu. Rev. Virol. 2018, 5, 453–476. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.M.; Armstrong, D.S.; Carzino, R.; Carlin, J.B.; Olinsky, A.; Robertson, C.F.; Grimwood, K. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J. Pediatr. 2001, 138, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.H.; Everett, J.; Trivedi, U.; Rumbaugh, K.P.; Whiteley, M. Requirement for pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 2014, 10, e1004518. [Google Scholar] [CrossRef] [PubMed]
- Mikel, P.; Vasickova, P.; Tesarik, R.; Malenovska, H.; Kulich, P.; Vesely, T.; Kralik, P. Preparation of MS2 phage-like particles and their use as potential process control viruses for detection and quantification of enteric RNA viruses in different matrices. Front. Microbiol. 2016, 7, 1911. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.A.; Richardson, C.C. Interactions of the RNA polymerase of bacteriophage T7 with its promoter during binding and initiation of transcription. Proc. Natl. Acad. Sci. USA 1986, 83, 3614–3618. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.N.; Fearns, R. How RNA viruses maintain their genome integrity. J. Gen. Virol. 2010, 91, 1373–1387. [Google Scholar] [CrossRef] [PubMed]
- Chamakura, K.; Young, R. Phage single-gene lysis: Finding the weak spot in the bacterial cell wall. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kidmose, R.T.; Vasiliev, N.N.; Chetverin, A.B.; Andersen, G.R.; Knudsen, C.R. Structure of the Qβ replicase, an RNA-dependent RNA polymerase consisting of viral and host proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 10884–10889. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Gaynor, J.B.; White, K.G.; Lopez, C.; Bosio, C.M.; Karkhoff-schweizer, R.R.; Schweizer, H.P. A tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2005, 2, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Bae, H.W.; Cho, Y.H. A pilin region affecting host range of the Pseudomonas aeruginosa RNA phage, PP7. Front. Micriobiol. 2018, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.T.; Niemela, S.L.; Miller, R.H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. USA 1989, 86, 2172–2175. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.G.; Urbach, J.M.; Wu, G.; Liberati, N.T.; Feinbaum, R.L.; Miyata, S.; Diggins, L.T.; He, J.; Saucier, M.; Déziel, E.; et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006, 7, R90. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G., III; Bloch, C.A.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; Gregor, J.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Chung, I.Y.; Choi, K.B.; Cho, Y.H. R-type pyocin is required for competitive growth advantage between Pseudomonas aeruginosa strains. J. Microbiol. Biotechnol. 2007, 17, 180–185. [Google Scholar]
- Heo, Y.J.; Ko, K.S.; Song, J.H.; Cho, Y.H. Profiling pyocins and competitive growth advantages in various Pseudomonas aeruginosa strains. J. Microbiol. Biotechnol. 2005, 15, 1368–1376. [Google Scholar]
- Kim, E.S.; Cho, Y.H. CHA University, Seongnam, Korea. Unpublished work. 2019. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-Y.; Ahn, S.-J.; Park, C.; Bae, H.-W.; Kim, E.S.; Cho, Y.-H. Reverse Genetic Systems for Pseudomonas aeruginosa Leviphages. Methods Protoc. 2019, 2, 22. https://doi.org/10.3390/mps2010022
Lee J-Y, Ahn S-J, Park C, Bae H-W, Kim ES, Cho Y-H. Reverse Genetic Systems for Pseudomonas aeruginosa Leviphages. Methods and Protocols. 2019; 2(1):22. https://doi.org/10.3390/mps2010022
Chicago/Turabian StyleLee, Jae-Yeol, Se-Jeong Ahn, Chanseop Park, Hee-Won Bae, Eun Sook Kim, and You-Hee Cho. 2019. "Reverse Genetic Systems for Pseudomonas aeruginosa Leviphages" Methods and Protocols 2, no. 1: 22. https://doi.org/10.3390/mps2010022
APA StyleLee, J.-Y., Ahn, S.-J., Park, C., Bae, H.-W., Kim, E. S., & Cho, Y.-H. (2019). Reverse Genetic Systems for Pseudomonas aeruginosa Leviphages. Methods and Protocols, 2(1), 22. https://doi.org/10.3390/mps2010022