Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors

 , ,
, ,
Abstract
1. Introduction
2. Experimental Design
2.1. Targeting Design and Human Umbilical Derived Erythroid Precursor 2 Cytogenetics
2.2. Materials
2.2.1. sgRNA Preparation
- pX458 & LKO1.5 (Addgene, Cambridge, MA, USA) [16]
- T4 Ligase 400U/μL & buffer (NEB, Ipswich, MA, USA; Cat. no.: M0202S).
- Oligonucleotides encoding sgRNA (Integrated DNA Technologies (IDT), Leuven, Belgium).
- BbsI FastDigest enzyme & buffer (ThermoFisher Scientific, Hemel Hempstead, UK; Cat. no.: FD1014).
- Midi Plasmid Plus Midi kit (Qiagen, Hilden, Germany; Cat. no.: 12943).
- Nuclease free water (Ambion, Foster City, CA, USA; Cat. no. AM9937).
- DH10B competent Escherichia coli bacteria (ThermoFisher Scientific; Cat. no.: 18297010).
- Surveyor Mutation Detection Kit (Integrated DNA Technologies; Cat. no. S100).
- jetPRIME transfection reagent (Polyplus-transfection, New York, NY, USA; Cat. no. 114-07).
- Proteinase K (Ambion; Cat. no.:2546).
- FastStart Taq DNA Polymerase deoxynucleotide triphosphates (dNTP) pack (Roche, Mannheim, Germany; Cat. no. 04738381001).
2.2.2. Growth Medium
- Stemspan SFEM (Stem Cell Technologies, Vancouver, BC, Canada; Cat. No. 09650).
- Glutamax 2 mM (ThermoFisher Scientific; Cat. no.: 25030-024).
- Penicillin-Streptomycin (100 units Penicillin and 100 μg Streptomycin/mL) (ThermoFisher; Cat. no.: 15140-122).
- Human Stem Cell Factor (SCF) (50 ng/mL) (Peprotech, Rocky Hill, NJ, USA; Cat. no: 300-07).
- Erythropoetin (3 IU/mL) (Janssen-Cilag, High Wycombe, UK, 10,000 IU/mL pre-filled syringe).
- Dexamethasone (DEX) (330 μg/L [840 nM]) (Hameln, Gloucester, UK; Cat. no.: DEXA3.3).
- Doxycycline (DOX) (1 μg/mL) (Sigma Aldrich, St Louis, MO, USA; Cat. no.: D3447).Note: Doxycycline has limited stability at 37 °C and requires supplementing every 24 h. Alternatively, 2 μg/mL doxycycline can be used in place of 1 μg/mL, enabling media to be changed every 48 h.
- HUDEP-2 cells were provided as an ongoing collaboration and cultured as described in Reference [8].
2.2.3. Freezing Medium
- 90% 0.4 μm filtered fetal bovine serum (Sigma Aldrich; Cat. no.: F7524).
- 10% dimethyl sulfoxide (DMSO) (Sigma Aldrich; Cat. no.: D2650).
2.2.4. Transfection
- 2B Amaxa Human CD34 Cell Nucleofector Kit (Lonza, Köln, Germany; Cat. no.: VPA-1003).
- RAD51-stimulatory compound-1 (RS-1) (Sigma Aldrich; Cat. no.: R9782).
- Complete media as described in Reference [8].
- Doxycycline (Sigma Aldrich; Cat. no.: D3447).
- Hoechst 33258 (Invitrogen, Carlsbad, CA, USA; Cat. no.: H3569).
2.3. Equipment
- AMAXA Nucleofector 2B (Amaxa, London, UK).
- Terasaki plates (Greiner, Kremsmünster, Austria; Cat no.: 653102).
- Fluorescence Activated Cell Sorter.
- Programmable Thermocycler.
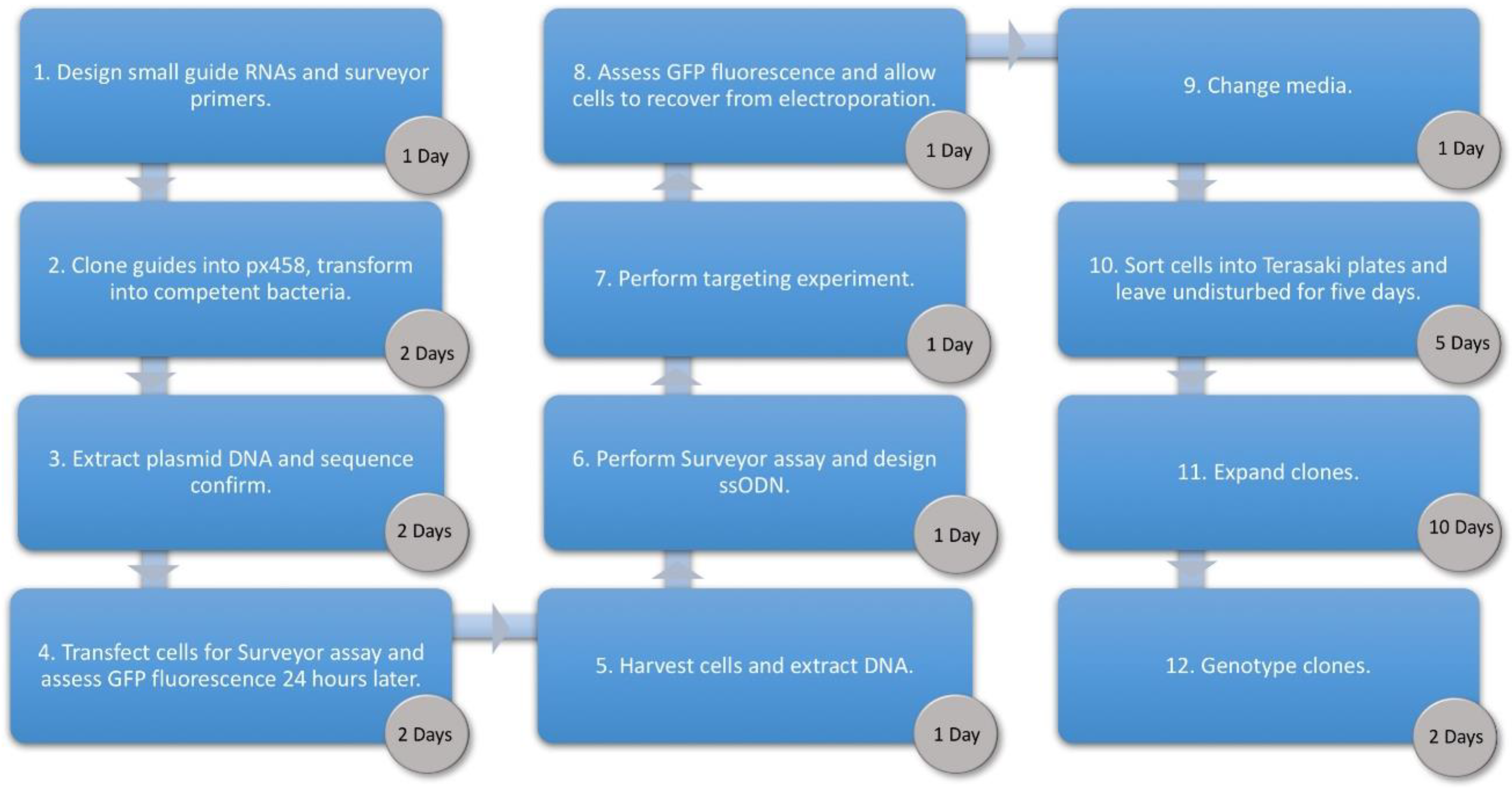
3. Procedure
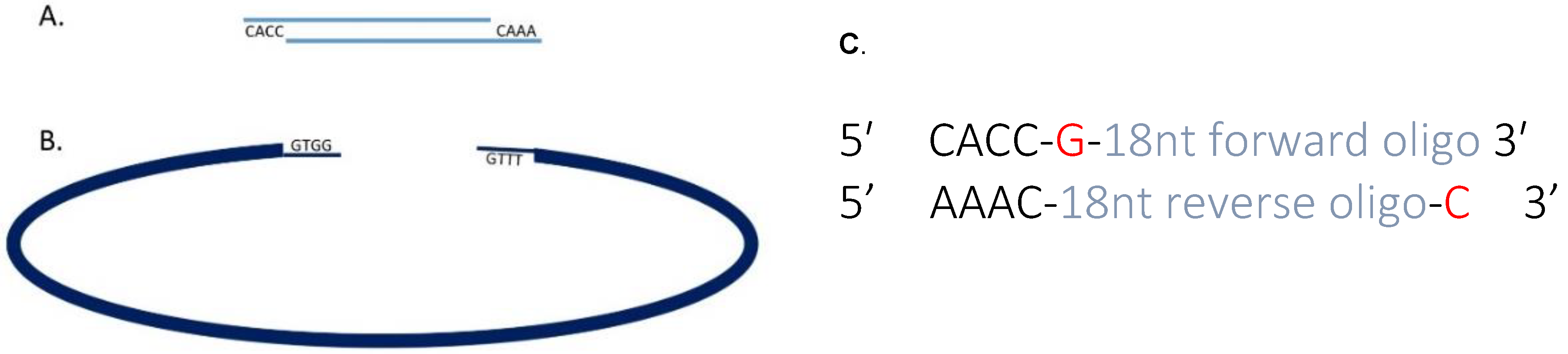
3.1. Cloning Oligonucleotides for Small Guide RNAs Time for Completion: 3 Days, Approx. 1 h per Day
- 1 μL of 100 μM sgRNA oligonucleotide F.
- 1 μL of 100 μM sgRNA oligonucleotide R.
- 8 μL nuclease free water.
- 1 μL BbsI 10× FastDigest buffer.
- 7.5 μL dH2O.
- 1 μL (0.5 μg) circular pX458 plasmid.
- 0.5 μL BbsI Fast Digest enzyme.
- 1.25 μL 10× T4 DNA Ligase Buffer.
- 0.5 μL heteroduplexed oligonucleotides.
- 0.75 μL Ligase.
- Total volume = 12.5 μL.
 CRITICAL STEP It is important to elute plasmids in a small volume (50 μL) to achieve concentrations of 4–6 μg/μL.
CRITICAL STEP It is important to elute plasmids in a small volume (50 μL) to achieve concentrations of 4–6 μg/μL.

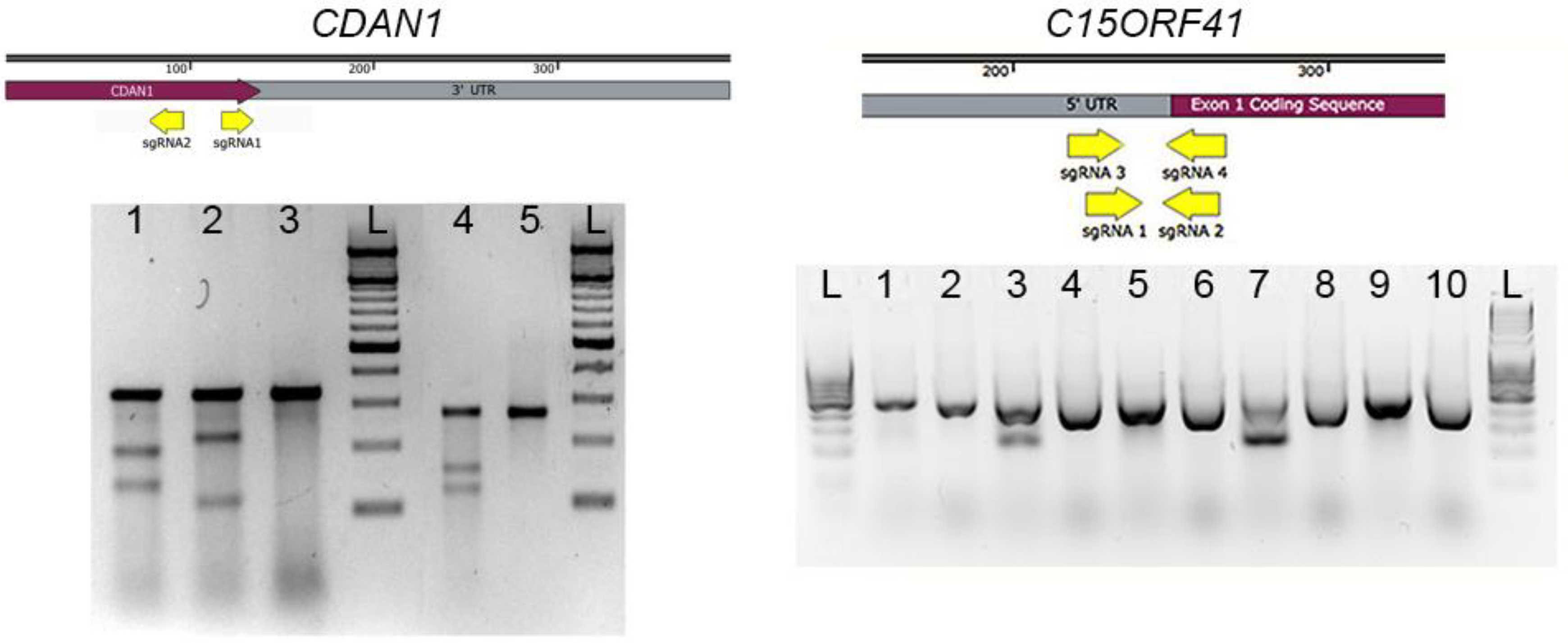
3.2. Surveyor Assay. Time for Completion: 1 Day
3.3. Design of Oligonucleotide Donor. Time for Completion: 2 h
3.4. Transfection of Human Umbilical Derived Erythroid Precursor 2 Cells. Time for Completion: 3 Days
3.4.1. Day 1. Time for Completion: 3 h
- 1.
- Count cells.
- 2.
- Aliquot 7.5 μg of plasmid, 9 μL 200 μM ssODN, 18.8 μL CD34 kit supplement and 81.2 μL CD34 kit buffer into a microcentrifuge tube.
 CRITICAL STEP The ratio of DNA to transfection reagents must be maintained as 1:10.
CRITICAL STEP The ratio of DNA to transfection reagents must be maintained as 1:10. CRITICAL STEP Do not exceed 110 μL total volume in the cuvette.
CRITICAL STEP Do not exceed 110 μL total volume in the cuvette.
- 3.
- Prepare the appropriate volume of media to resuspend cells at a density of 1 × 106 per mL with 2 μg/mL DOX.
- 4.
- Aliquot 3.25 × 106 cells into a tube and centrifuge at 270× g for 5 min.
- 5.
- Aspirate supernatant, resuspend the cells by flicking the tube then add >10mL of phosphate buffered saline (PBS) and centrifuge at 270× g for 5 min.
- 6.
- Aspirate PBS removing as much liquid as possible. This is important in ensuring an efficient transfection.
- 7.
- Resuspend cells pellets in buffer/supplement/plasmid/donor solution and transfer (<110 μL) into a Nucleofector Amaxa 2B cuvette by gently dispensing the cells down the side of the vessel between the metallic plates and without introducing any bubbles.
- 8.
- Nucleofect the cells by placing the cuvette in the Amaxa 2B Nucleofector using protocol U-08.
- 9.
- Immediately following nucleofection return the cuvette to tissue culture hood and add growth media (see Section 2.2 for description) containing RS-1 (if required) to the cuvette. Aim to dilute the buffer a minimum of 5:1 media:buffer but ideally 10:1 within the first minute post nucleofection. Keep 1 mL of resuspension volume to wash cuvette with after transferring the cells into a culture vessel.
- 10.
- Using the Pasteur pipette provided in the nucleofection kit, remove cells from the cuvette and gently triturate the solution to evenly distribute them.
- 11.
- Rinse the cuvette with the remaining 1 mL of media and add this to the culture vessel.
- 12.
- Resuspend the cells to a density of 1 × 106 cells/mL in the prewarmed media with 2 μg/mL DOX
- OPTIONAL STEP If you are attempting to integrate a donor at the cut site it is possible to use the small molecule RS-1 to promote homology driven repair (0.75 μM final concentration in the cell resuspension media) [21,22]. RS-1 is a small molecule activator of RAD51, which is thought to be involved in finding a homologous repair template and facilitating strand exchange [23].
3.4.2. Day 2. Time for Completion: 20 min
- 13.
- Assess cells for fluorescence using a microscope. Expect 30–50% GFP-positive cells.
3.4.3. Day 3. Time for Completion: 30 min
- 14.
- Count cells and perform a full media change by centrifuging cells at 270× g for five minutes and resuspending cells in growth medium at a density of 1 × 106 cells/mL. RS-1 is no longer required.
3.5. Cell Sorting. Time for Completion: 6 h
Day 4
- 15.
- Using fluorescence activated cell sorting (FACS) sort single cells into each well of a 60-well microtiter plate (Terasaki plate). Each well should be prefilled with 20 μL media containing 2 μg/mL DOX and maintained in an incubator until the cells have been prepared for sorting.
- 16.
- Prepare both the transfected cells and 1 × 105 untransfected cells by centrifugation at 270× g for five minutes and washing once with PBS. After recentrifugation and discarding PBS, resuspend the cell pellet in complete media with 2 μg/mL DOX and 1 μg/mL Hoechst 33258 to a density of 1 × 106–5 × 106 per mL as appropriate depending on the FACS machine and preferred flow rate. Hoechst is used to differentiate live/dead cells during sorting.
- 17.
- Set gate for GFP-positive cells based on the untransfected population and sort single GFP-positive cells into each well of 10 Terasaki plates.
- 18.
- Once cells have been sorted, transfer Terasaki plates to a humidity box (any container that allows gas exchange but retains moisture) and place in an incubator.
3.6. Clone Expansion. Time for Completion: 14 Days
3.6.1. Days 5–9
3.6.2. Day 10
- 19.
- Take note of which wells have cells in them using an inverted light microscope. Clones that have cell populations that cover more than a third of the well should be transferred into a 96 well plate in 100 μL of media. Approximately 25–35% of sorted cells expand enough to be transferred.
- 20.
- Clones that have cover less than one third of the well should be left in situ until they have further expanded and add a dilution of doxycycline (dilute 1 mg/mL 1:50 and add 2 μL to the 20 μL for 2 μg/mL final concentration). Mix gently, no more than four times, with a pipette set to 15 μL and visually confirm cells have been evenly redistributed in the well. Clones that have not expanded after 15 days post-sort should be discarded (unless slow growth is a likely outcome relevant to the target locus in which case further time may be given).Note: If the wells contain less than 15 μL, first add the appropriate volume of complete media.
3.6.3. Day 11 onwards
- 21.
- Expand the clones from 20 μL Terasaki wells → 100 μL 96 well → 200 μL 96well → 2× 200 μL 96 wells → 500 μL in 24 well → 1 mL in 24 well. Expect ~30 clones to survive this process.
- 22.
- Once the cells are confluent in 1 mL of media in the 24 well plate, prepare 1.5 mL microcentrifuge tubes labelled with a unique ID for each clone and an equivalent cryovial.
- 23.
- For each well, gently mix the cell suspension to evenly distribute the cells, then aliquot 500 μL into the cryovial and 500 μL into the matched microcentrifuge tube.
- 24.
- Add 500 μL of freezing media to the cryovial and mix by pipetting up and down once before sealing the cap and submerging the vial in a container of dry ice.
- 25.
- Add 1 mL of PBS to the microcentrifuge tube and spin at 350× g for 5 min.
- 26.
- Aspirate as much PBS as possible from the pelleted cells and then submerge the tube into dry ice.
- 27.
- Cryovials should be stored at −80 °C for up to four weeks and transferred to liquid or vapour phase nitrogen cryostorage thereafter.
- 28.
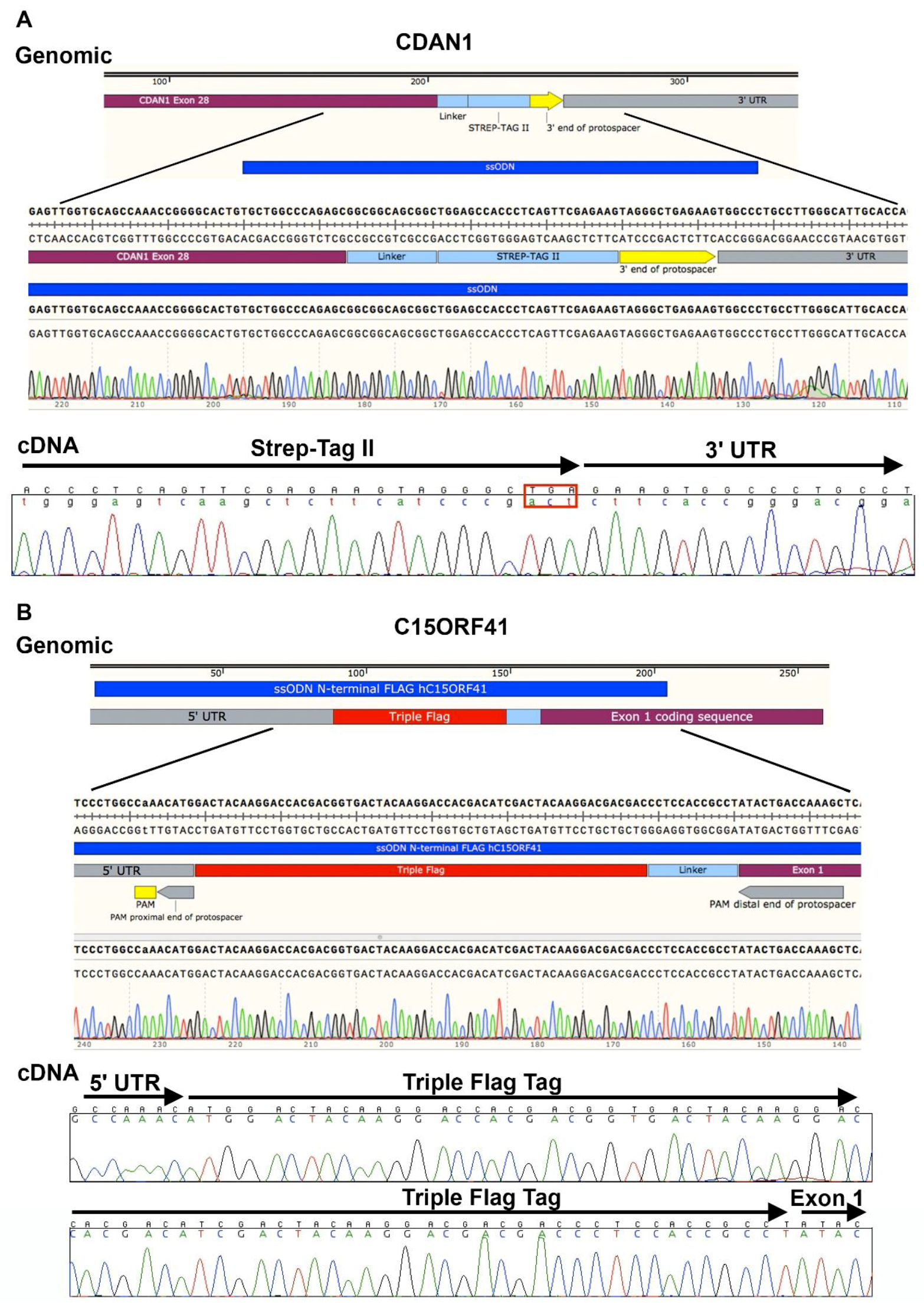
- DNA can be extracted from the cell pellets (as described in Section 3.2) in microcentrifuge tubes and screened by polymerase chain reaction (PCR) amplification and Sanger sequencing coupled with TIDER software [24] or restriction digest to determine correct integration of the required genomic modification. Indels may be analyzed in by combining Sanger sequencing with TIDE software [25], or amplification products may be cloned and sequenced. Next generation sequencing (NGS) is extremely useful in the analysis of HDR and indels and a novel method describing high-throughput multiplexed screening of modified clones is reported in Nussbaum et al. [26].
- 29.
- Recover the correctly targeted clones in 2× 200 μL 96 wells and check them 24 h post-thaw.
- OPTIONAL STEP Clones that are growing poorly can be supplemented with 2× EPO (6 IU/mL), 2× SCF (100 ng/mL) and 2× DEX (1.68 μM) (as well as 2 μg/mL DOX) every second day until they are in 2× 96 well.
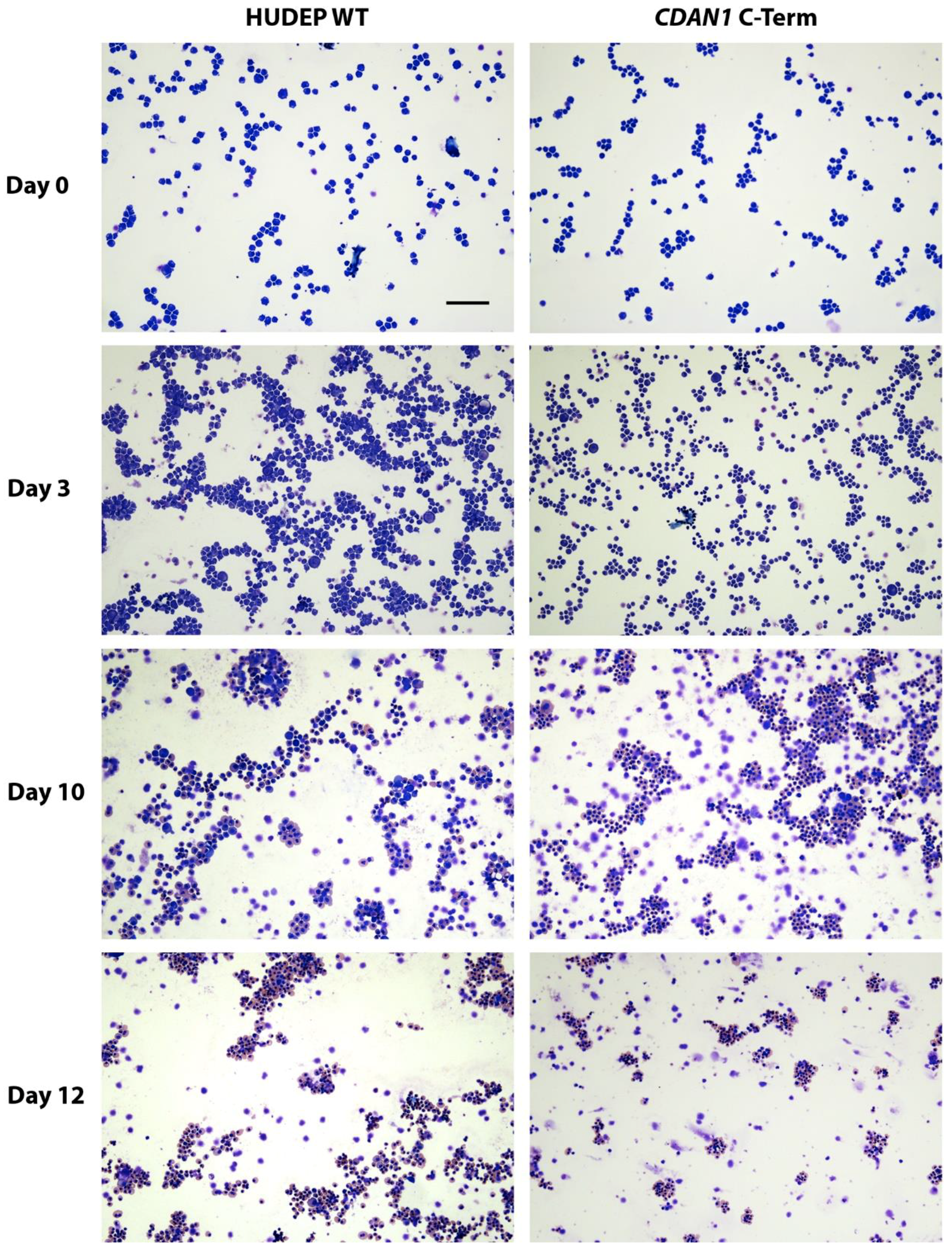
4. Expected Results
Author Contributions
Funding
Conflicts of Interest
Appendix A. aCGH Analysis
References
- Lozzio, C.B.; Lozzio, B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 1975, 45, 321–334. [Google Scholar] [PubMed]
- Martin, P.; Papayannopoulou, T. HEL cells: A new human erythroleukemia cell line with spontaneous and induced globin expression. Science 1982, 216, 1233–1235. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Rifkind, R.A. Erythroleukemic differentiation. Annu. Rev. Biochem. 1978, 47, 419–448. [Google Scholar] [CrossRef] [PubMed]
- Cigudosa, J.C.; Odero, M.D.; Calasanz, M.J.; Solé, F.; Salido, M.; Arranz, E.; Martínez-Ramirez, A.; Urioste, M.; Alvarez, S.; Cervera, J.V.; et al. De novo erythroleukemia chromosome features include multiple rearrangements, with special involvement of chromosomes 11 and 19. Genes Chromosomes Cancer 2003, 36, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Trakarnsanga, K.; Wilson, M.C.; Lau, W.; Singleton, B.K.; Parsons, S.F.; Sakuntanaga, P.; Kurita, R.; Nakamura, Y.; Anstee, D.J.; Frayne, J. Induction of adult levels of beta-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica 2014, 991, 677–685. [Google Scholar]
- Zurlo, J.; Rudacille, D.; Goldberg, A.M. The three Rs: The way forward. Environ. Health Perspect. 1996, 104, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Khoriaty, R.; Vasievich, M.P.; Jones, M.; Everett, L.; Chase, J.; Tao, J.; Siemieniak, D.; Zhang, B.; Maillard, I.; Ginsburg, D. Absence of a red blood cell phenotype in mice with hematopoietic deficiency of SEC23B. Mol. Cell. Biol. 2014, 34, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Kurita, R.; Sudo, K.; Suda, N.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 2013, 8, e59890. [Google Scholar]
- Wienert, B.; Martyn, G.E.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 2017, 130, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Congenital Dyserythropoietic Anemia Type I. GeneReviews®. University of Washington, Seattle, Washington, DC, USA, 1993. Posted 21 April 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK5313/ (accessed on 18 July 2018).
- Babbs, C.; Roberts, N.A.; Sanchez-Pulido, L.; McGowan, S.J.; Ahmed, M.R.; Brown, J.M.; Sabry, M.A.; Bentley, D.R.; McVean, G.A.; Donnelly, P.; et al. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia Type I. Haematologica 2013, 98, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, M.A.; Corn, J.E.; Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods 2017, 121, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. CRISPR/Cas9 Genome editing in human hematopoietic stem cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed]
- University of California. UCSC Genome Browser, BLAT Search Genome. Available online: https://genome.ucsc.edu/cgi-bin/hgBlat?command=start (accessed on 18 July 2018).
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Goomer, R.S.; Kunkel, G.R. The Transcriptional Start Site for a Human U6 Small Nuclear RNA Gene is Dictated by a Compound Promoter Element Consisting of the PSE and the TATA Box. Nucleic Acids Res. 1992, 20, 4903–4912. [Google Scholar] [CrossRef] [PubMed]
- Cost, G.J. Enzymatic ligation assisted by nucleases: Simultaneous ligation and digestion promote the ordered assembly of DNA. Nat. Protoc. 2007, 2, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.G.; Koepke, K.; Frank, R.; Skerra, A. Molecular interaction between the strep-tag affinity peptide and its cognate target, streptavidin. J. Mol. Biol. 1996, 255, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Hopp, T.P.; Prickett, K.S.; Price, V.L.; Libby, R.T.; March, C.J.; Cerretti, D.P.; Urdal, D.L.; Conlon, P.J. A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat. Biotechnol. 1988, 6, 1204–1210. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-Mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [PubMed]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Kousholt, A.N.; Harmsen, T.; Leemans, C.; Chen, T.; Jonkers, J.; van Steensel, B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018, 46, e58. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Armendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, L.; Telenius, J.M.; Hill, S.; Hirschfeld, P.P.; Suciu, M.C.; The WIGWAM Consortium; Downes, D.J.; Hughes, J.R. High-Throughput Genotyping of CRISPR/Cas Edited Cells in 96-Well Plates. Methods Protoc. 2018, 1, 29. [Google Scholar]
- Vinjamur, D.S.; Bauer, D.E. Growing and genetically manipulating human umbilical cord blood-derived erythroid progenitor (HUDEP) cell lines. Methods Mol. Biol. 2018, 1698, 275–284. [Google Scholar] [PubMed]
- Trakarnsanga, K.; Griffiths, R.E.; Wilson, M.C.; Blair, A.; Satchwell, T.J.; Meinders, M.; Cogan, N.; Kupzig, S.; Kurita, R.; Nakamura, Y.; et al. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 2017, 8, 14750. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Homology-directed repair of DNA Nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, e924–e932. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Li, X.L.; Chen, W. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Prediger, E. A Recombinant Cas9 Enzyme That Drastically Reduces CRISPR Off-Target Effects. Integrated DNA Technologies. Available online: https://eu.idtdna.com/pages/education/decoded/article/a-recombinant-cas9-enzyme-that-drastically-reduces-crispr-off-target-effects (accessed on 18 July 2018).
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konerman, S. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.X.; Hansen, L.L.; Artiles, K.L.; Nonet, M.L.; Fire, A.Z. Landscape of target:guide homology effects on Cas9-mediated cleavage. Nucleic Acids Res. 2014, 42, w13778–w13787. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reynon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.I.; Kim, J.S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.; Fuller, C.K.; Donohouse, P.D.; Jones, B.N.; Thompson, M.S.; Carter, M.M.; Gradia, S.; Vidal, B.; Garner, E.; Slorach, E.M.; et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods 2017, 14, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.; Reutzel, D.; Speicher, M. Complete karyotype characterization of the K562 cell line by combined application of G-banding, multiplex-fluorescence in situ hybridization, fluorescence in situ hybridization, and comparative genomic hybridization. Leuk. Res. 2001, 25, 313–322. [Google Scholar] [CrossRef]
- Lessard, S.; Gatof, E.S.; Beaudoin, M.; Schupp, P.G.; Sher, F.; Ali, A.; Prehar, S.; Kurita, R.; Nakamura, Y.; Baena, E.; et al. An erythroid-specific ATP2B4 enhancer mediates red blood cell hydration and malaria susceptibility. J. Clin. Investig. 2017, 127, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Canver, M.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Lessard, S.; Pinello, L.; Wu, Y.; Ilboudo, Y.; Stern, E.N.; Needleman, A.J.; Galactéros, F.; Brugnara, C.; Kutlar, A.; et al. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat. Genet. 2017, 49, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Traxler, E.A.; Tao, Y.; Wang, Y.D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Database of Genomic Variants. Available online: http://dgv.tcag.ca/dgv/app/home (accessed on 18 July 2018).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Start (hg18 *) | End (hg18) | Event | Length | Cytoband | Count of Genes |
|---|---|---|---|---|---|---|
| 3 | 46,990,391 | 50,479,675 | ROH | 3,489,285 | p21.31 | 121 |
| 3 | 156,276,396 | 159,447,085 | ROH | 3,170,690 | q25.31–q25.33 | 24 |
| 6 | 0 | 171,115,067 | CN Gain | 171,115,068 | p25.3–q27 | 1,433 |
| 8 | 0 | 83,821,458 | CN Gain | 83,821,459 | p23.3–q21.13 | 597 |
| 8 | 83,821,459 | 110,503,487 | CN Gain | 26,682,029 | q21.13–q23.2 | 165 |
| 8 | 110,503,488 | 125,424,290 | CN Gain | 14,920,803 | q23.2–q24.13 | 65 |
| 8 | 125,424,291 | 138,306,862 | CN Gain | 12,882,572 | q24.13–q24.23 | 76 |
| 8 | 138,306,863 | 146,364,022 | CN Gain | 8,057,160 | q24.23–q24.3 | 141 |
| 14 | 37,393,981 | 37,510,754 | CN Loss | 116,774 | q13.3 | 2 |
| 17 | 30,301,223 | 81,195,210 | CN Gain | 50,893,988 | q11.2–q25.3 | 1,022 |
| 18 | 0 | 15,375,878 | CN Loss | 15,375,879 | p11.32–p11.21 | 119 |
| 18 | 18,561,020 | 78,077,248 | CN Gain | 59,516,229 | q11.1–q23 | 310 |
| 19 | 0 | 59,128,983 | CN Gain | 59,128,984 | p13.3–q13.43 | 1,872 |
| 21 | 14,368,320 | 48,129,895 | CN Gain | 33,761,576 | q11.2–q22.3 | 363 |
| Locus | % Alleles Repaired by NHEJ | % Alleles Targeted by HDR |
|---|---|---|
| CDAN1 C-Term | 80% | 20% |
| C15ORF41 N-Term | 65% | 35% |
| C15ORF41 C-Term | 70% | 30% |
| ATRX | 58% | 42% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moir-Meyer, G.; Cheong, P.L.; Olijnik, A.-A.; Brown, J.; Knight, S.; King, A.; Kurita, R.; Nakamura, Y.; Gibbons, R.J.; Higgs, D.R.; et al. Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors. Methods Protoc. 2018, 1, 28. https://doi.org/10.3390/mps1030028
Moir-Meyer G, Cheong PL, Olijnik A-A, Brown J, Knight S, King A, Kurita R, Nakamura Y, Gibbons RJ, Higgs DR, et al. Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors. Methods and Protocols. 2018; 1(3):28. https://doi.org/10.3390/mps1030028
Chicago/Turabian StyleMoir-Meyer, Gemma, Pak Leng Cheong, Aude-Anais Olijnik, Jill Brown, Samantha Knight, Andrew King, Ryo Kurita, Yukio Nakamura, Richard J. Gibbons, Douglas R. Higgs, and et al. 2018. "Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors" Methods and Protocols 1, no. 3: 28. https://doi.org/10.3390/mps1030028
APA StyleMoir-Meyer, G., Cheong, P. L., Olijnik, A.-A., Brown, J., Knight, S., King, A., Kurita, R., Nakamura, Y., Gibbons, R. J., Higgs, D. R., Buckle, V. J., & Babbs, C. (2018). Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors. Methods and Protocols, 1(3), 28. https://doi.org/10.3390/mps1030028