A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies

Abstract
1. Introduction
2. Experimental Design
2.1. Materials
- pET-NLS-Cas9-6xHis was a gift from David Liu (Addgene, Cambridge, MA, USA; plasmid no. 62934)
- Escherichia coli BL21 star (DE3)pLysS (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: C602003)
- High vacuum grease (Dow Corning, Midland, MI, USA; Cat. no.: 1597418)
- HEPES (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: H-3375)
- Potassium chloride (Sigma-Aldrich; Cat. no.: P3911)
- Magnesium chloride hexahydrate (Sigma-Aldrich; Cat. no.: M2670)
- Sodium chloride (Sigma-Aldrich; Cat. no.: S7653)
- Tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Sigma-Aldrich; Cat. no.: C4706)
- Imidazole (Sigma-Aldrich; Cat. no.: I-2399)
- Glycerol (Sigma-Aldrich; Cat. no.: G5516)
- Ni-NTA superflow (Qiagen, Hilden, Germany; Cat. no.: 30430)
- Econo-Pac chromatography columns (Bio-Rad; Hercules, CA, USA; Cat. no.: 7321010)
- Protein assay dye reagent concentrate (Bio-Rad; Hercules, CA, USA; Cat. no.: 5000006)
2.2. Equipment
- French pressure cell (35 mL cell with 1” diameter piston; AMINCO, Silver Spring, MD, USA; Cat. no.: 4-3339)
- French pressure cell press (AMINCO, Lake Forest, CA, USA)
- Beckman J2-MI centrifuge (Beckman, Brea, CA, USA)
- Beckman Coulter Ja-17 rotor (Beckman)
- Beckman Coulter Allegra X-15R centrifuge (Beckman)
- Beckman Coulter SX4750 swinging bucket rotor (Beckman)
- Resource S, 6 mL cation exchange column (GE Healthcare, Uppsala, Sweden; Cat. no.: 17-1180-01)
- ӒKTA FPLC system (Amersham Biosciences, Little Chalfont, UK)
- Amicon Ultra-15 centrifugal filters, 50K cutoff (Millipore, Burlington, MA, USA; Cat. no.: UFC905024)
- Spartan-30 HPLC (High-performance liquid chromatography) certified syringe filter with regenerated cellulose membrane, diameter: 30 mm, pore size: 0.2 μM (Whatman, Maidstone, UK; Cat. no.: 10463060).
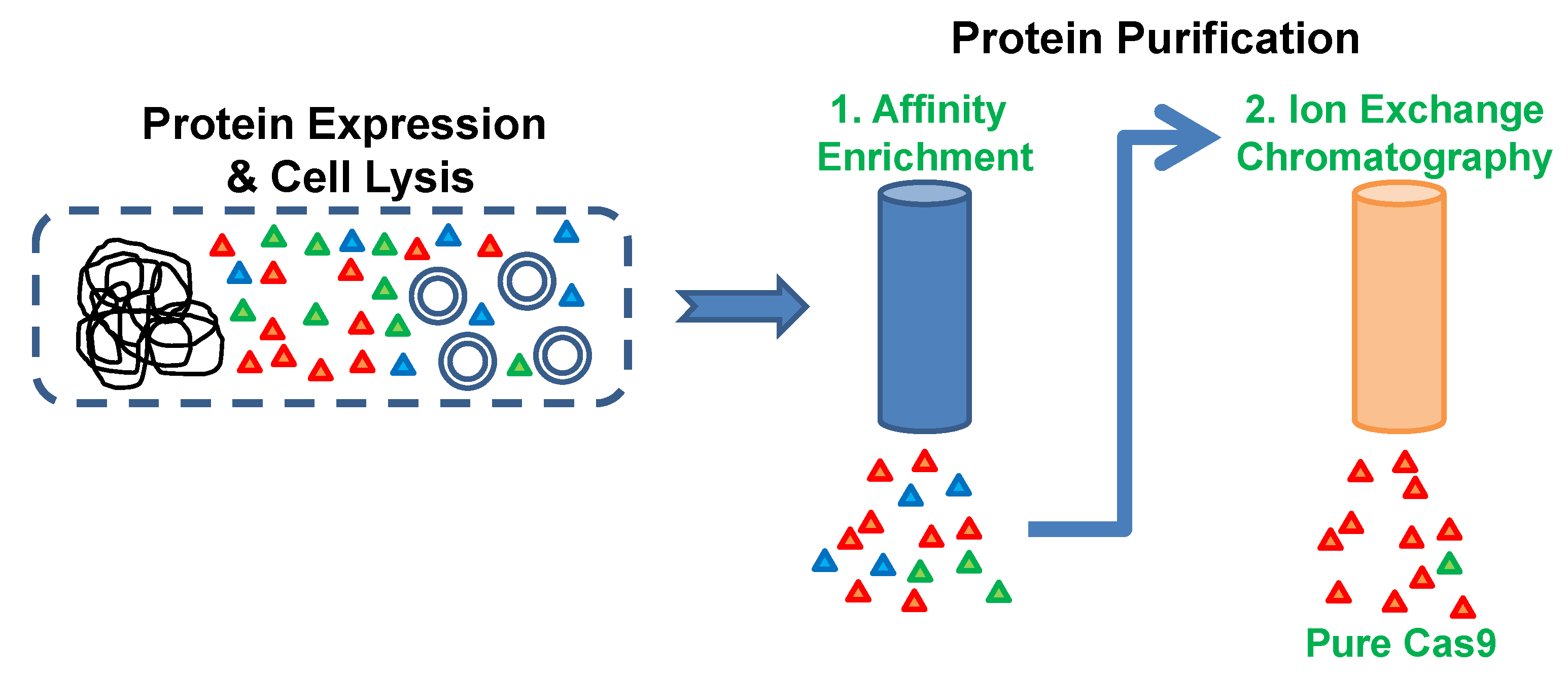
3. Procedure
3.1. Cell Lysis and Isolation of Soluble Proteins. Time to Completion: 1 h
- Adjust the volume of harvested cells to 35 mL by adding ice-cold buffer LW (for cell lysis and washing out unbound molecules).
- Suspend the cell mass in the buffer thoroughly by vortexing and placing the tube in an end-over-end rotation shaker. To prevent clogging of the cell lysis equipment, ensure that there are no cell clumps at the end of this step.
- Load the cell suspension into a pre-cooled 35 mL French pressure cell equipped with a 1-inch diameter piston. Lubricate the piston and cap of the French pressure cell with high vacuum grease to ensure smooth operation.
- Close the cell and load it in the cell press device. Set the cell pressure to approximately 16,000 psi and open the key gently to release the suspension and collect the cell lysate in a fresh tube. The key should not be opened wide and the cell pressure should be maintained close to the set pressure.
- Repeat steps 3 and 4 to ensure proper lysis of the cell suspension.
- Separate the cell debris by centrifuging the lysate at 39,706× g at 4 °C for 20 min.
- Quickly decant the supernatant into a fresh tube and proceed to metal affinity purification with this sample.
- OPTIONAL STEP Filter the supernatant through a 0.22 μM syringe filter to ensure the removal of any cells from the pellet.
3.2. Metal Affinity Chromatography. Time for Completion: 2 h
- Take 3 mL of Ni-NTA affinity resin suspension in a 20 mL gravity flow chromatography column.
- Let the storage buffer flow through and wash the affinity resin thrice with 4 mL of ice-cold buffer LW.
- Add the washed affinity resin (by suspending it in a small volume of buffer LW) to the cell lysis supernatant from step 3.1.
 CRITICAL STEP Incubate this reaction tube in an end-over-end rotating shaker for exactly 15 min. As we do not add any protease inhibitors to the purification buffers, it is important to minimize the length of this incubation.
CRITICAL STEP Incubate this reaction tube in an end-over-end rotating shaker for exactly 15 min. As we do not add any protease inhibitors to the purification buffers, it is important to minimize the length of this incubation.- Load the suspension on a 20 mL gravity flow chromatography column and let the unbound fraction flow through.
- Wash the resin thrice with 4 mL of ice-cold buffer LW.
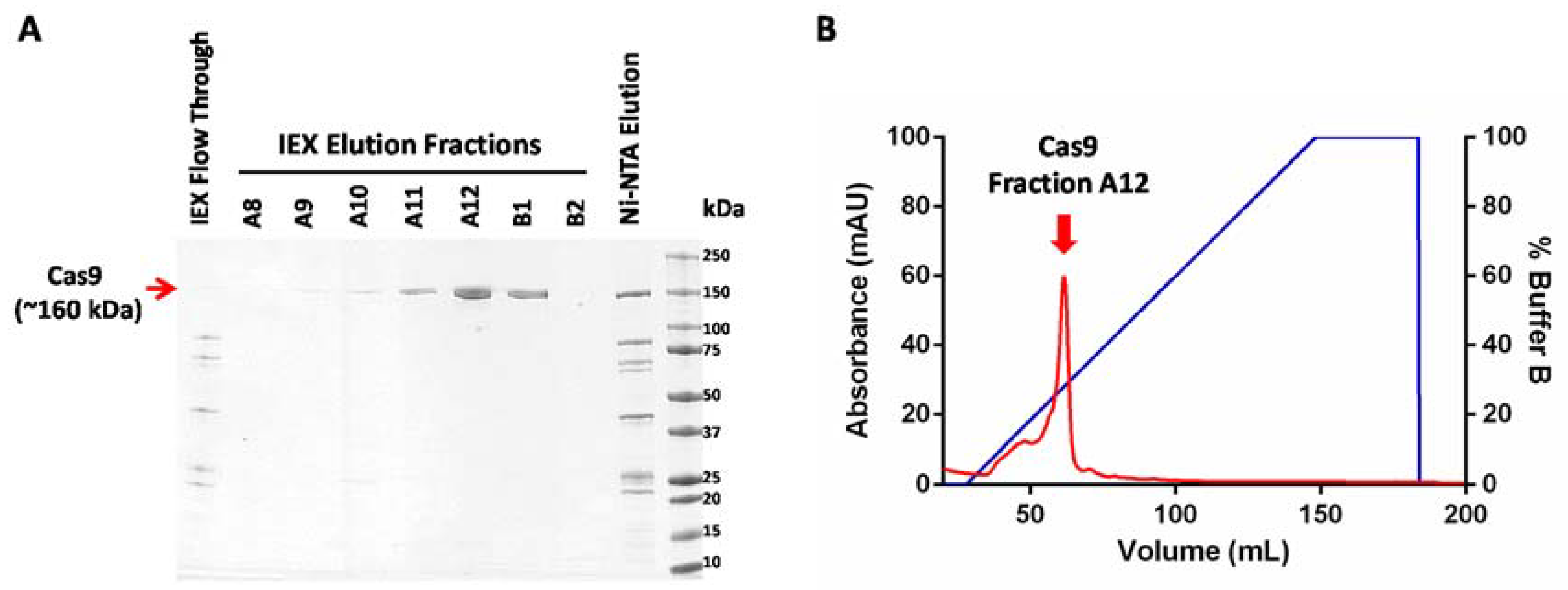
- Elute bound Cas9 from the column by adding ice-cold buffer EB (elution buffer) and collecting eight 1.5 mL fractions.
- Estimate the concentration of protein in the elution fractions using an appropriate protein assay method (e.g., Bradford protein assay).
 OPTIONAL PAUSE STEP The elution fractions with the highest protein concentrations can be pooled and dialyzed overnight against 1 liter of buffer A at 4 °C using a 50,000 Da or lower molecular weight cutoff membrane. While this step might serve as a pause point, it can be replaced by diluting the sample with buffer A or conducting buffer exchange using a centrifugal filter device to complete the entire protein purification within a day.
OPTIONAL PAUSE STEP The elution fractions with the highest protein concentrations can be pooled and dialyzed overnight against 1 liter of buffer A at 4 °C using a 50,000 Da or lower molecular weight cutoff membrane. While this step might serve as a pause point, it can be replaced by diluting the sample with buffer A or conducting buffer exchange using a centrifugal filter device to complete the entire protein purification within a day.


3.3. Cation Exchange Chromatography. Time to Completion: 1 h
- OPTIONAL STEP The pooled elution fractions from step 3.2 can be concentrated and buffer exchanged into buffer A using a centrifugal filter. However, this optional step could add 30 min to the protocol.
- For a chromatography system connected with a 10 mL sample injection loop, the pooled elution fractions can be diluted to 10 mL using buffer A. A two times dilution of the sample with buffer A was sufficient to enable its binding to the column.
 CRITICAL STEP Filter the sample by passing it through a 0.2 μM syringe filter before loading onto the chromatography column.NOTE: To save time and avoid degradation of protein, all the chromatography steps described below can be performed at the maximum permissible flow rate of 6 mL/minute for this column.
CRITICAL STEP Filter the sample by passing it through a 0.2 μM syringe filter before loading onto the chromatography column.NOTE: To save time and avoid degradation of protein, all the chromatography steps described below can be performed at the maximum permissible flow rate of 6 mL/minute for this column.- Inject the sample into a Resource S (6 mL) cation exchange chromatography column that is pre-equilibrated with 5 column volumes (CV) (30 mL) of buffer A.
- Wash unbound proteins and buffer EB salts using 2 CV (12 mL) of buffer EB.
- Elute bound proteins by increasing the volume of buffer B from 0 to 100% over 20 CV (120 mL). Collect 5 mL fractions of the elution.
- Wash column with 5 CV of 100% buffer B followed by re-equilibration with 5 CV of 100% buffer A.
- Estimate the concentration of protein in the elution peaks using the Bradford protein assay reagent.
3.5. Buffer Exchange and Protein Concentration. Time for Completion: 1 h
- Pool together fractions from the Cas9 elution peak (as indicated in Figure 2) and concentrate it using a centrifugal filter with a molecular weight cutoff limit of 50,000. The swinging bucket rotor can be set at a maximum of 4000× g for these filters and the samples can be spun for 5 min at 4 °C.
- Once a roughly 10-times concentration is achieved, dilute the sample to the original volume with buffer A and centrifuge for a further 5 min.
- Repeat this step at least three times to exchange the sample into buffer A.
- Adjust the final volume of the sample according to the desired protein concentration for the downstream application.
 PAUSE STEP Add glycerol to the protein sample and aliquot into small usable fractions. Freeze samples with liquid nitrogen and store them at −80 °C. A final concentration of 5–20% glycerol can be used depending on the downstream application. These frozen samples can be stored for several months. However, repeated freeze–thaw cycles of the same fractions should be avoided.
PAUSE STEP Add glycerol to the protein sample and aliquot into small usable fractions. Freeze samples with liquid nitrogen and store them at −80 °C. A final concentration of 5–20% glycerol can be used depending on the downstream application. These frozen samples can be stored for several months. However, repeated freeze–thaw cycles of the same fractions should be avoided.
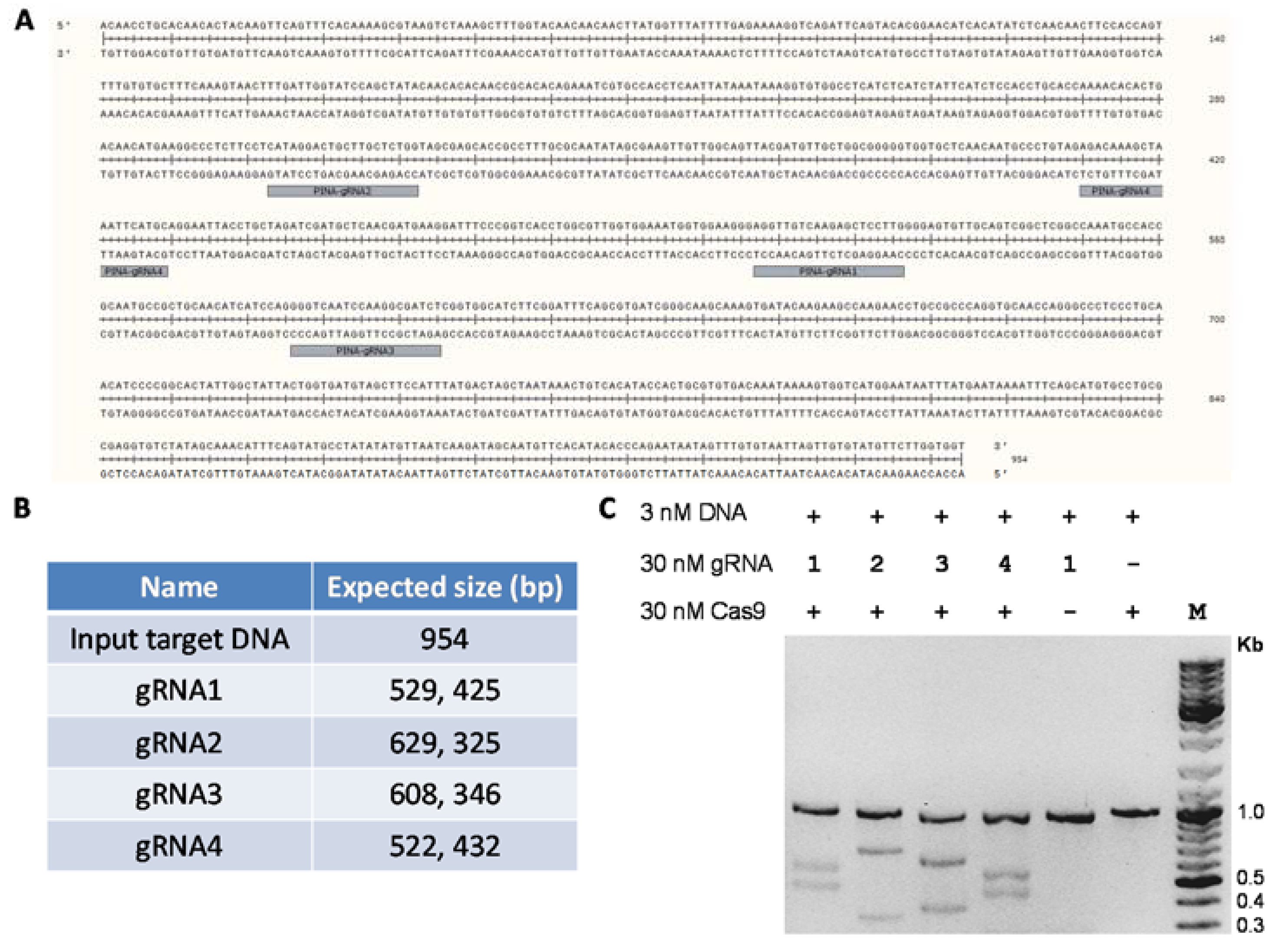
4. Expected Results
5. Reagents Setup
- Unless stated otherwise, all reagents were prepared with autoclaved dd H2O (henceforth, referred to as water)
- 1 M HEPES pH 7.5. Dissolve 119.15 g of HEPES in 300 mL water, set pH with NaOH solution to 7.5, adjust final volume to 500 mL with water, filter sterilize through a 0.22 µM filter and store at room temperature for several weeks.
- 2 M KCl. Dissolve 149.1 g of KCl in 1 L of water, sterilize by autoclaving and store at room temperature for several weeks.
- 1 M MgCl2. Dissolve 50.8 g of MgCl2·6H2O in 250 mL water, sterilize by autoclaving and store at room temperature for several weeks.
- 3 M NaCl. Dissolve 175.32 g of NaCl in 1 L of water, sterilize by autoclaving and store at room temperature for several weeks.
- 2.5 M Imidazole. Dissolve 1.7 g of imidazole in 10 mL of water, filter sterilize through a 0.22 µM filter and store at 4 °C for a week.
- 0.5 M TCEP. Dissolve 1.43 g of TCEP in 10 mL of water, filter sterilize through a 0.22 µM filter and store at 4 °C for several weeks.
- Buffer LW (for cell lysis, affinity column equilibration and washing unbound proteins).20 mM HEPES pH 7.5, 300 mM NaCl, 25 mM Imidazole, 0.5 mM TCEP. Mix together 2 mL of 1 M HEPES pH 7.5, 10 mL 3 M NaCl, 1 mL 2.5 M imidazole, 0.5 M TCEP, check pH and adjust to 7.5 using HCl, adjust final volume to 100 mL with water and sterilize by passing through a 0.22 μM filter and store at 4 °C for up to a week.
- Buffer EB (for elution from affinity column).20 mM HEPES pH 7.5, 300 mM NaCl, 250 mM Imidazole, 0.5 mM TCEP. Mix together 1 mL of 1 M HEPES pH 7.5, 5 mL of 3 M NaCl, 5 mL of 2.5 M imidazole, 0.05 mL of 0.5 M TCEP, check pH and adjust to 7.5 using HCl, adjust final volume to 50 mL with water and sterilize by passing through a 0.22 μM filter and store at 4 °C for up to a week.
- Buffer A (for cation exchange column equilibration and washing unbound proteins).20 mM HEPES pH 7.5, 200 mM KCl, 10 mM MgCl2, 0.5 mM TCEP. Mix together 8 mL of 1 M HEPES pH 7.5, 40 mL of 2 M KCl, 4 mL of 1 M MgCl2, 0.4 mL of 0.5 M TCEP, adjust final volume to 400 mL with water, sterilize and degas by passing through a 0.22 μM filter connected to a vacuum pump. Store at 4 °C for up to a week.
- Buffer B (for elution of proteins from cation exchange column).20 mM HEPES pH 7.5, 1 M KCl, 10 mM MgCl2, 0.5 mM TCEP. Mix together 4 mL of 1 M HEPES pH 7.5, 100 mL of 2 M KCl, 2 mL of 1 M MgCl2, 0.2 mL of 0.5 M TCEP, adjust final volume to 200 mL using water, sterilize and degas by passing through a 0.22 μM filter connected to a vacuum pump. Store at 4 °C for up to a week.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucl. Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Yang, Y.; Wang, N.; Shui, S.; Kim, S.; Kanchiswamy, C.N.; Barbas, C.F.; Liu, J.; Kim, J.-S. Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat. Protoc. 2015, 10, 1842–1859. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, K.; Zhang, Y.; Liu, J.; Yin, K.; Qiu, J.; Gao, C. Genome editing of bread wheat using biolistic delivery of CRISPR/Cas9 in vitro transcripts or ribonucleoproteins. Nat. Protoc. 2018, 13, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2014, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample Name | Concentration (mg/mL) | Volume (mL) | Total Protein (mg) |
|---|---|---|---|
| Metal affinity enrichment | |||
| Ni-NTA Elution 1 | 5.9 | 1.5 | 8.85 |
| Ni-NTA Elution 2 | 9.8 | 1.5 | 14.7 |
| Ni-NTA Elution 3 | 1.2 | 1.5 | 1.8 |
| Ni-NTA Elution 4 | 0.3 | 1.5 | 0.45 |
| Ni-NTA Elution 5 | 0.2 | 1.5 | 0.3 |
| Ni-NTA Elution 6 | 0.1 | 1.5 | 0.15 |
| Ni-NTA Elution 7 | 0.1 | 1.5 | 0.15 |
| Ni-NTA Elution 8 | 0 | 1.5 | 0 |
| Ni-NTA Total | 26.4 | ||
| Cation exchange chromatography | |||
| IEX A11 | 0.04 | 5 | 0.2 |
| IEX A12 | 0.43 | 5 | 2.15 |
| IEX B1 | 0.1 | 5 | 0.5 |
| IEX Total | 2.85 | ||
| Centrifugal filter buffer exchange and concentration | |||
| Final Cas9 yield | 2.4 | 1 | 2.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajagopalan, N.; Kagale, S.; Bhowmik, P.; Song, H. A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies. Methods Protoc. 2018, 1, 17. https://doi.org/10.3390/mps1020017
Rajagopalan N, Kagale S, Bhowmik P, Song H. A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies. Methods and Protocols. 2018; 1(2):17. https://doi.org/10.3390/mps1020017
Chicago/Turabian StyleRajagopalan, Nandhakishore, Sateesh Kagale, Pankaj Bhowmik, and Halim Song. 2018. "A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies" Methods and Protocols 1, no. 2: 17. https://doi.org/10.3390/mps1020017
APA StyleRajagopalan, N., Kagale, S., Bhowmik, P., & Song, H. (2018). A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies. Methods and Protocols, 1(2), 17. https://doi.org/10.3390/mps1020017