A Fluorometric Method for the Quantification of Cell Number in Complex Differentiating Osteoblast-Osteocyte Cultures


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Equipment
2.2. Experimental Procedure
2.2.1. Sample Preparation
2.2.2. Stabilization of Cells
2.2.3. Generation of Cell Lysates
2.2.4. GelRedTM Staining on Genomic DNA
2.2.5. Fluorescence Intensity Reading and Cell Number Determination
3. Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, P.; Henning, S.M.; Heber, D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS ONE 2010, 5, e10202. [Google Scholar] [CrossRef] [PubMed]
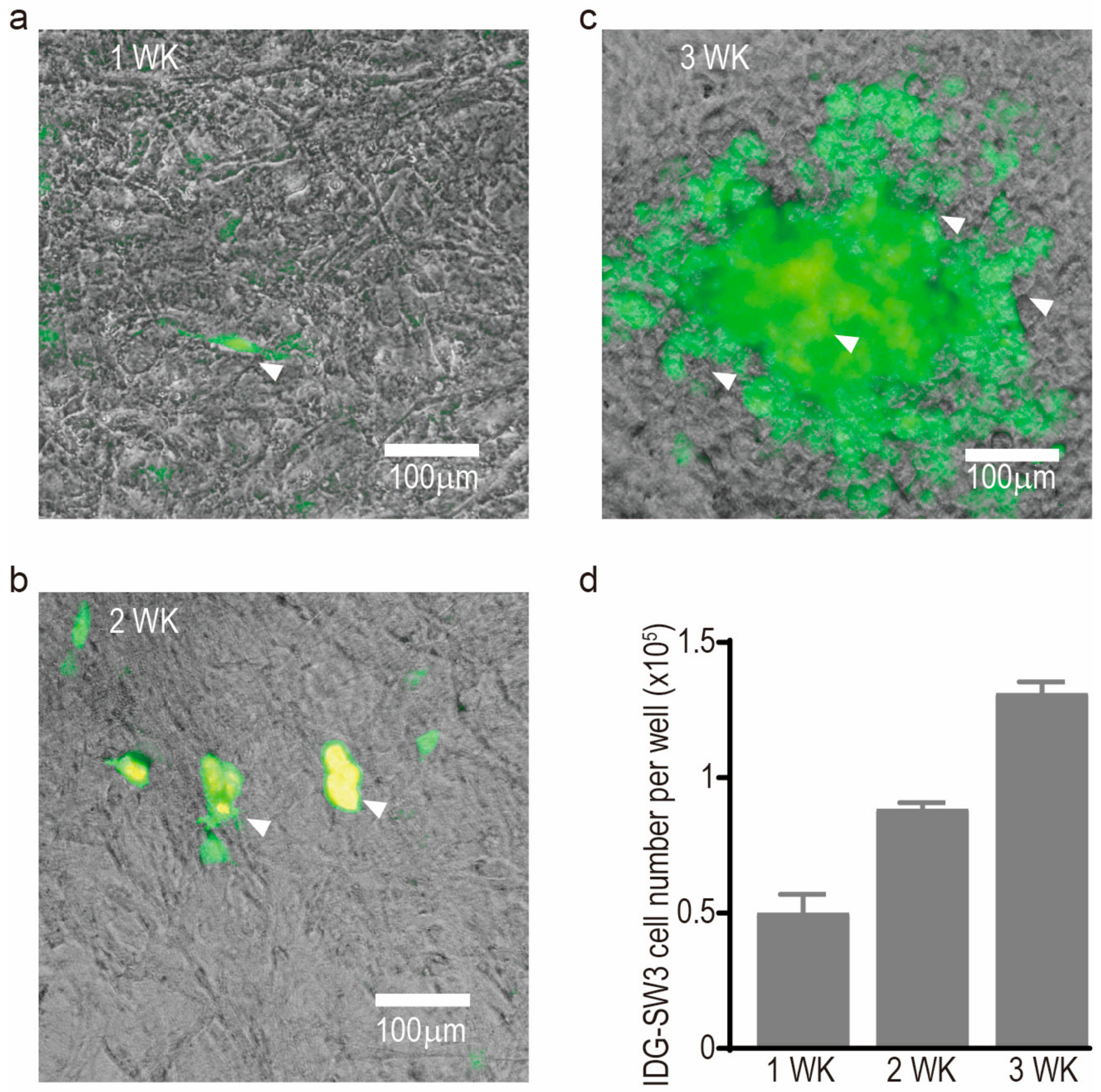
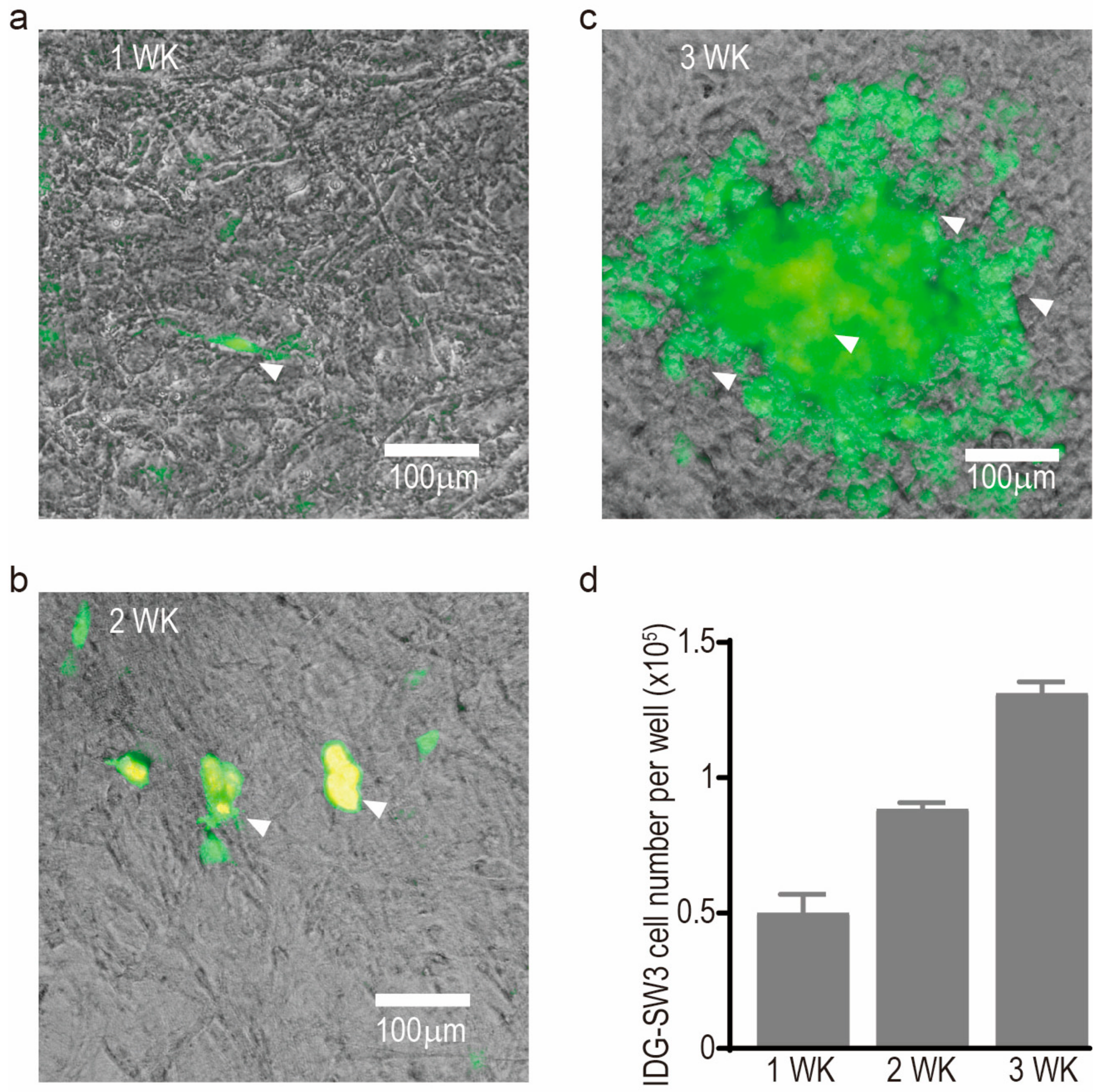
- Woo, S.M.; Rosser, J.; Dusevich, V.; Kalajzic, I.; Bonewald, L.F. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J. Bone Miner. Res. 2011, 26, 2634–2646. [Google Scholar] [CrossRef] [PubMed]
- Coxon, F.P. Fluorescence imaging of osteoclasts using confocal microscopy. Methods Mol. Biol. 2012, 816, 401–424. [Google Scholar] [PubMed]
- Tenenbaum, H.C.; Heersche, J.N. Differentiation of osteoblasts and formation of mineralized bone in vitro. Calcif. Tissue Int. 1982, 34, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N Y Acad. Sci. 2010, 1192, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Boskey, A.; Spevak, L.; Dallas, M.; Hori, M.; Bonewald, L.F. Establishment of an osteoid preosteocyte-like cell MLO-A5 that spontaneously mineralizes in culture. J. Bone Miner. Res. 2001, 16, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.J.; Kostakis, P.; Pan, B.; Farrugia, A.; Gronthos, S.; Evdokiou, A.; Harrison, K.; Findlay, D.M.; Zannettino, A.C. RANKL expression is related to the differentiation state of human osteoblasts. J. Bone Miner. Res. 2003, 18, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
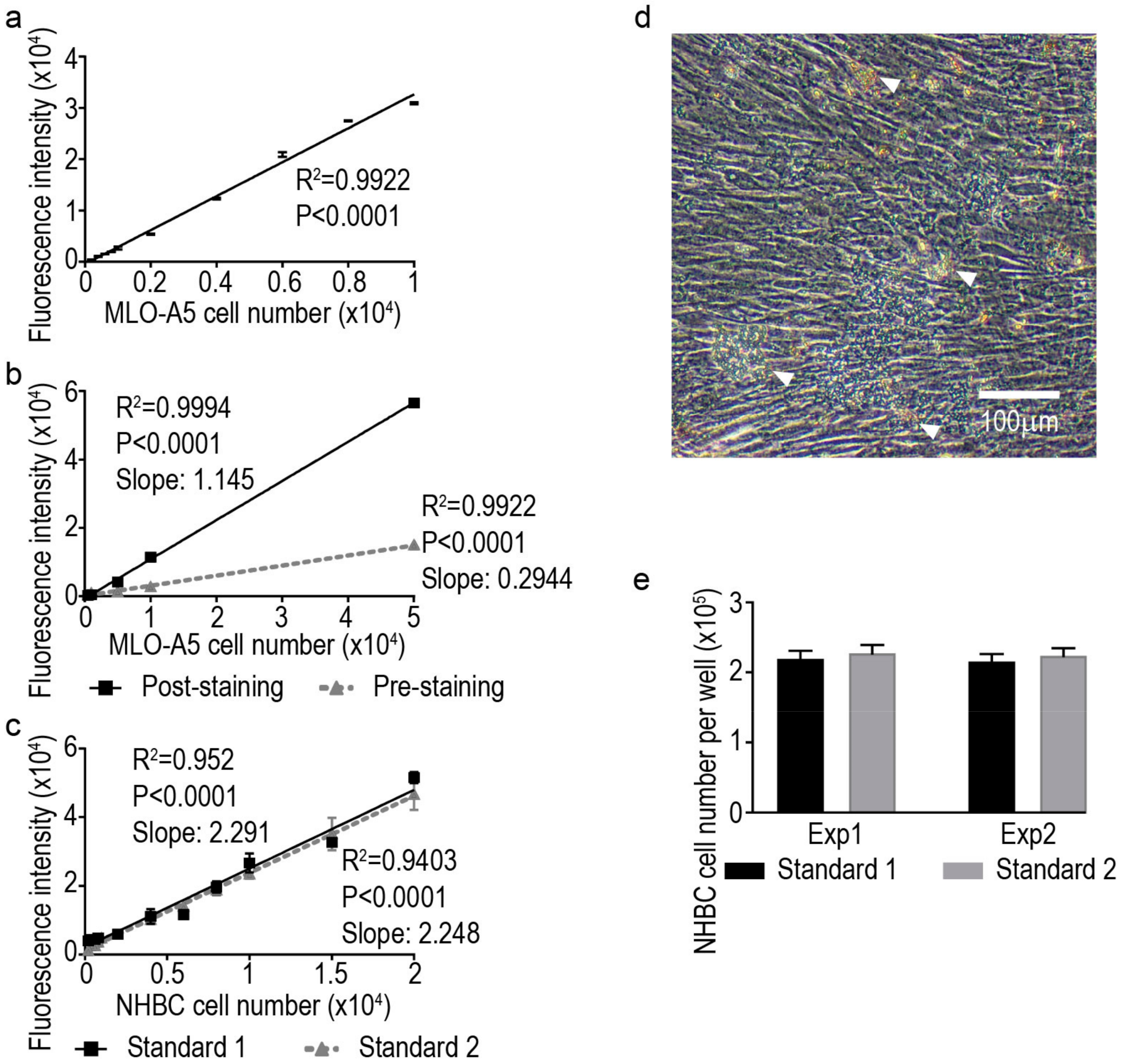
 ) and pre-staining (
) and pre-staining (  ) methods; (c) standard curves generated from human primary bone-derived cells (NHBC) suspension cultures from two independent experiments; (d) representative image of a differentiated NHBC culture under phase-contrast with high level of confluence and deposited mineral nodules (indicated by arrowheads); and (e) cell number measurements from two independent experiments by returned calculation using the two standard curves above, respectively. Each bar graph represents the average cell number from four independent culture wells and the error bar represents the standard error of the mean.
) and pre-staining ( ) methods; (c) standard curves generated from human primary bone-derived cells (NHBC) suspension cultures from two independent experiments; (d) representative image of a differentiated NHBC culture under phase-contrast with high level of confluence and deposited mineral nodules (indicated by arrowheads); and (e) cell number measurements from two independent experiments by returned calculation using the two standard curves above, respectively. Each bar graph represents the average cell number from four independent culture wells and the error bar represents the standard error of the mean.
) methods; (c) standard curves generated from human primary bone-derived cells (NHBC) suspension cultures from two independent experiments; (d) representative image of a differentiated NHBC culture under phase-contrast with high level of confluence and deposited mineral nodules (indicated by arrowheads); and (e) cell number measurements from two independent experiments by returned calculation using the two standard curves above, respectively. Each bar graph represents the average cell number from four independent culture wells and the error bar represents the standard error of the mean.
) and pre-staining ( ) methods; (c) standard curves generated from human primary bone-derived cells (NHBC) suspension cultures from two independent experiments; (d) representative image of a differentiated NHBC culture under phase-contrast with high level of confluence and deposited mineral nodules (indicated by arrowheads); and (e) cell number measurements from two independent experiments by returned calculation using the two standard curves above, respectively. Each bar graph represents the average cell number from four independent culture wells and the error bar represents the standard error of the mean.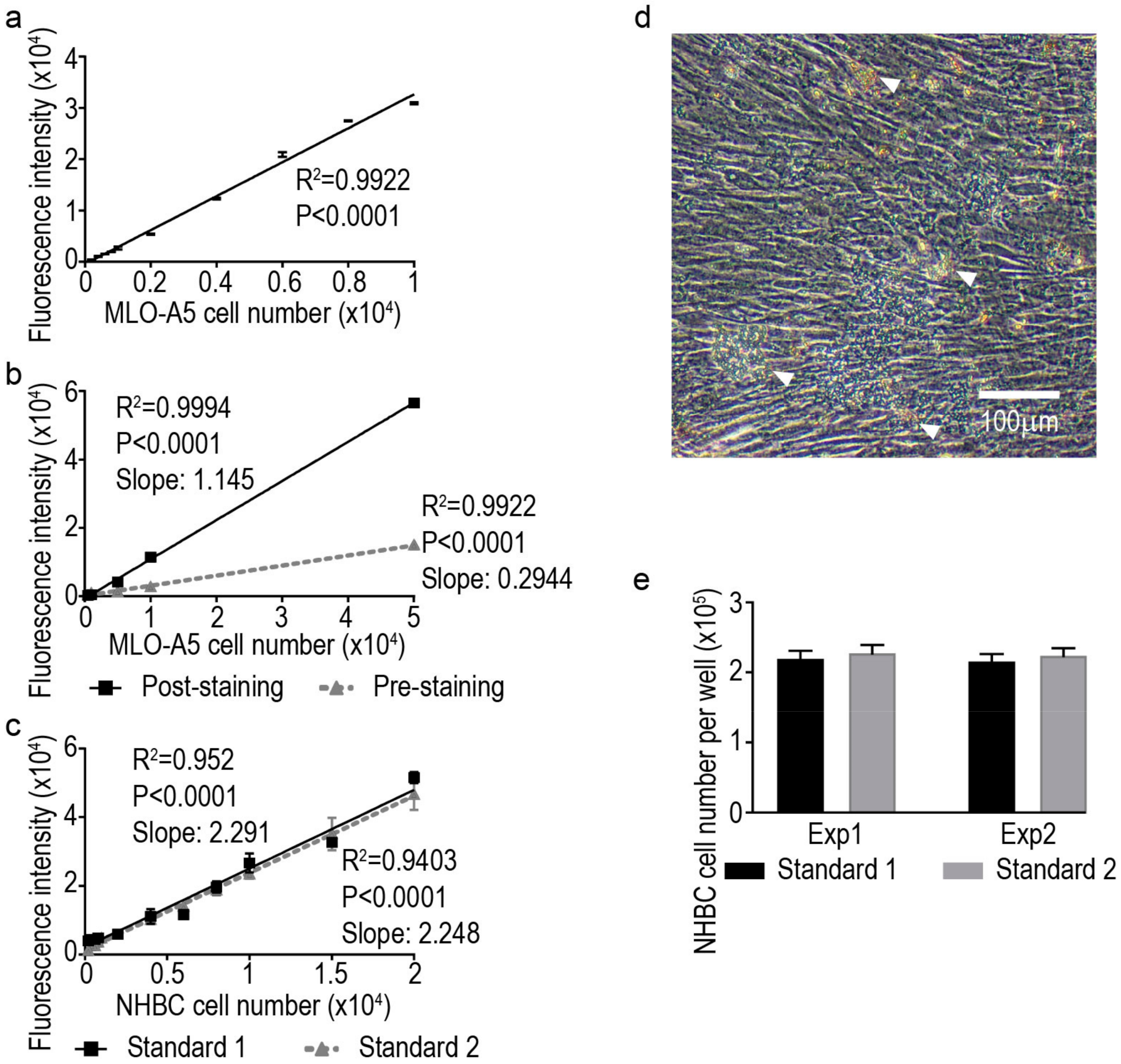
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Wijenayaka, A.R.; Atkins, G.J. A Fluorometric Method for the Quantification of Cell Number in Complex Differentiating Osteoblast-Osteocyte Cultures. Methods Protoc. 2018, 1, 14. https://doi.org/10.3390/mps1020014
Yang D, Wijenayaka AR, Atkins GJ. A Fluorometric Method for the Quantification of Cell Number in Complex Differentiating Osteoblast-Osteocyte Cultures. Methods and Protocols. 2018; 1(2):14. https://doi.org/10.3390/mps1020014
Chicago/Turabian StyleYang, Dongqing, Asiri R. Wijenayaka, and Gerald J. Atkins. 2018. "A Fluorometric Method for the Quantification of Cell Number in Complex Differentiating Osteoblast-Osteocyte Cultures" Methods and Protocols 1, no. 2: 14. https://doi.org/10.3390/mps1020014
APA StyleYang, D., Wijenayaka, A. R., & Atkins, G. J. (2018). A Fluorometric Method for the Quantification of Cell Number in Complex Differentiating Osteoblast-Osteocyte Cultures. Methods and Protocols, 1(2), 14. https://doi.org/10.3390/mps1020014