
A Newborn Screening Program for Sickle Cell Disease in Murcia (Spain)

 , , ,
, , , 
Abstract
:1. Introduction
2. Objective
3. Methodology
3.1. Subjects
3.2. Hemoglobin Variant Analysis
4. Results
5. Discussion
6. Limitations and Strengths
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erythropathology Group of the Spanish Society of Haematology and Haemotherapy. Guide to Sickle Cell Disease; Medea, Medical Education Agency S.L.: Madrid, Spain, 2021. [Google Scholar]
- Brandow, A.M.; Liem, R.I. Advances in the diagnosis and treatment of sickle cell disease. J. Hematol. Oncol. 2022, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Lees, C.M.; Davies, S.; Dezateux, C. Neonatal screening for sickle cell disease. Cochrane Database Syst. Rev. 2000, 2000, CD001913. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.B.; Mehal, J.M.; Chapman, C.; Haberling, D.L.; Richardson, L.C.; Bean, C.J.; Hooper, W.C. Trends in sickle cell disease-related mortality in the United States, 1979 to 2017. Ann. Emerg. Med. 2020, 76, S28–S36. [Google Scholar] [CrossRef] [PubMed]
- Sobota, A.; Sabharwal, V.; Fonebi, G.; Stibleinberg, M. How we prevent and manage infection in sickle cell disease. Br. J. Haematol. 2015, 170, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.J.; Madu, A.; Sangeda, R.Z.; Nkya, S.; Peprah, E.; Paintsil, V.; Mmbando, B.P.; Gyamfi, J.; Okocha, C.E.; Asala, S.A.; et al. Utilization of Pneumococcal Vaccine and Penicillin Prophylaxis in Sickle Cell Disease in Three African Countries: Assessment among Healthcare Providers in SickleInAfrica. Hemoglobin 2021, 45, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.H.; Verter, J.I.; Woods, G.; Pegelow, C.; Kelleher, J.; Presbury, G.; Zarkowsky, H.; Vichinsky, E.; Iyer, R.; Lobel, J.S.; et al. Prophylaxis with oral penicillin in children with sickle cell anemia. N. Engl. J. Med. 1986, 314, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hurst, D.; Earles, A.; Kleman, K.; Lubin, B. Newborn screening for sickle cell disease: Effect on mortality. Pediatrics 1988, 81, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Thomas, P.; Cupidore, L.; Serjeant, B.; Serjeant, G. Improved survival in homozygous sickle cell disease: Lessons from a cohort study. BMJ 1995, 311, 1600–1602. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for the Control of Haemoglobin Disorders. 1994. Available online: https://apps.who.int/iris/bitstream/handle/10665/66665/WHO_HDP_HB_GL_94.1.pdf (accessed on 15 February 2019).
- Health Resources and Services Administration. Newborn Screening: Toward a Uniform Screening Panel and System. 2015. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/newborn-uniform-screening-panel.pdf (accessed on 3 July 2018).
- UK National Screening Committee. NHS Sickle Cell & Thalassaemia Screening Programme: Standards for the Linked Antenatal and Newborn Screening Programme; NHS Sickle Cell and Thlassaemia Screening Programme Center: London, UK, 2011. [Google Scholar]
- Población Según Zonas Básicas de Salud, Municipio y Área de Salud. Portal Estadístico de la Región de Murcia. 2023. Available online: https://econet.carm.es/inicio//crem/sicrem/PU_padron/p20/sec29.html#:~:text=CREM%20%20PADR%C3%93N%20MUNICIPAL%20DE%20HABITANTES%20%2026.,salud.%202020%20Fecha%20de%20actualizaci%C3%B3n%3A%2004%2F02%2F2021.%20-%20CREM (accessed on 3 June 2023).
- Cancho, E.J.B.; García-Morín, M.; Beléndez, C.; Velasco, P.; Benéitez, D.; Ruiz-Llobet, A.; Berrueco, R.; Argilés, B.; Cervera, Á.; Salinas, J.A.; et al. Update of the Spanish registry of haemoglobinopathies in children and adults. Med. Clin. 2020, 155, 95–103. [Google Scholar]
- Núñez-Jurado, D.; Payán-Pernía, S.; Álvarez-Ríos, A.I.; Jiménez-Jambrina, M.; Pérez-De-Soto, I.C.; Palma-Vallellano, A.J.; Zapata-Bautista, R.; Hernández-Castellet, J.C.; Garrastazul-Sánchez, M.P.; Arqueros-Martínez, V.; et al. Neonatal Screening for Sickle Cell Disease in Western Andalusia: Results and Lessons Learnt after 3 Years of Implementation. Am. J. Perinatol. 2022. online ahead of print. [Google Scholar]
- Culliton, B.J. Genetic screening: NAS recommends proceeding with caution. Science 1975, 189, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Wethers, D.; Pearson, H.; Gaston, M. Newborn screening for sickle cell disease and other hemoglobinopathies. JAMA 1987, 258, 1205–1209. [Google Scholar] [CrossRef]
- Shafer, F.E.; Lorey, F.; Cunningham, G.C.; Klumpp, C.; Vichinsky, E.; Lubin, B. Newborn screening for sickle cell disease: 4 yearsof experience from California’s newborn screening program. J. Pediatr. Hematol. Oncol. 1996, 83, 813–814. [Google Scholar] [CrossRef] [PubMed]
- Panepinto, J.A.; Magid, D.; Rewers, M.J.; Lane, P.A. Universalversus targeted screening of infants for sickle cell disease: A cost-effectiveness analysis. J. Pediatr. 2000, 136, 201–208. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
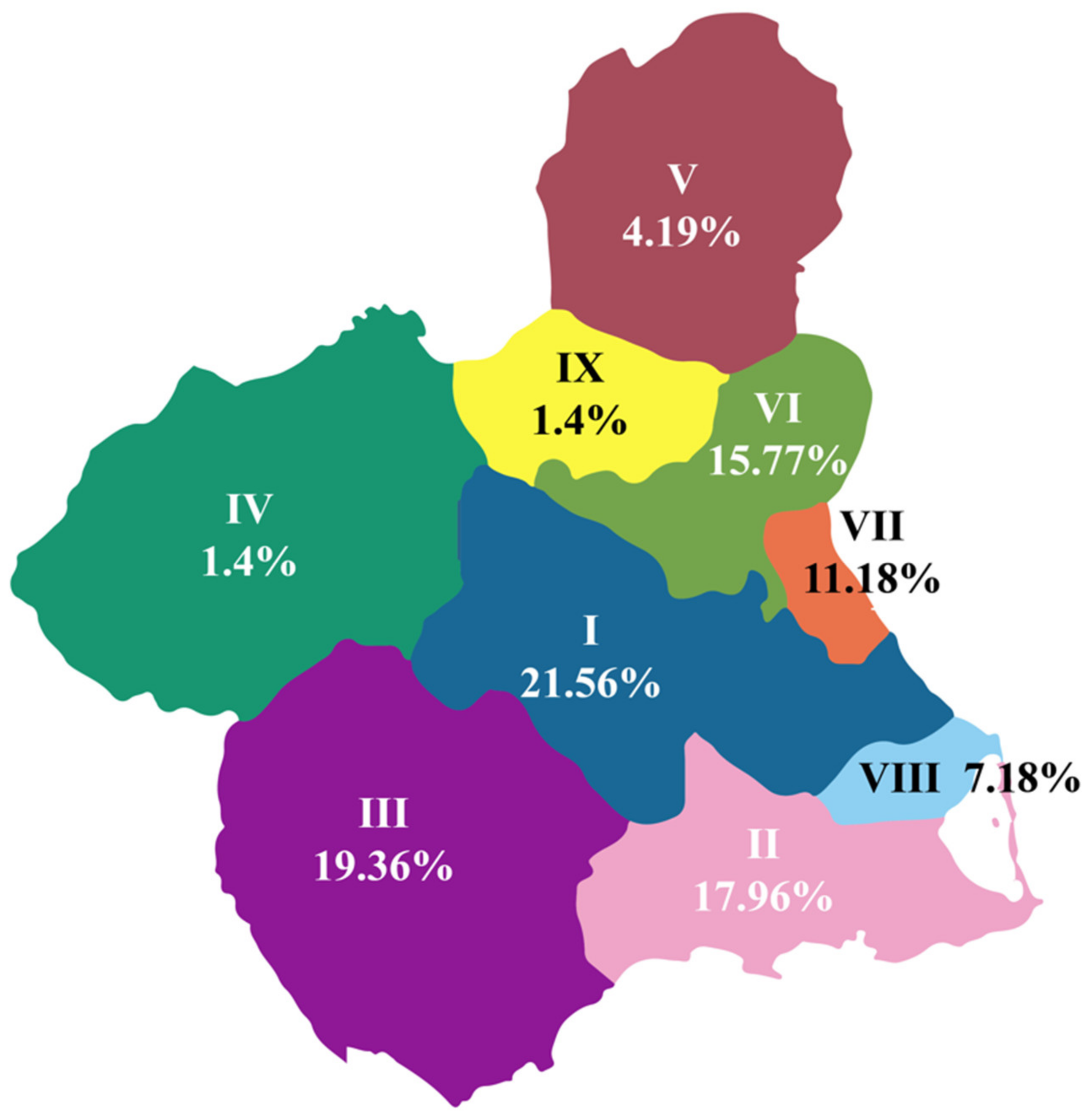
| Health Area | % | Total Cases | Population [13] | Incidence (per 100,000) |
|---|---|---|---|---|
| I | 21.56 | 108.0156 | 269,627 | 4.01 |
| II | 17.96 | 89.9796 | 288,536 | 3.12 |
| III | 19.36 | 96.9936 | 180,577 | 5.37 |
| IV | 1.4 | 7.014 | 69,947 | 1.00 |
| V | 4.19 | 20.9919 | 60,828 | 3.45 |
| VI | 15.77 | 79.0077 | 272,042 | 2.90 |
| VII | 11.18 | 56.0118 | 204,969 | 2.73 |
| VIII | 7.18 | 36.0219 | 109,851 | 3.28 |
| IX | 1.4 | 7.014 | 54,874 | 1.28 |
| Total | 100 | 501 | 1,511,251 | |
| Average Incidence | 3.02 |
| Hemoglobin Variant | Number (%) |
|---|---|
| Sickle Cell Syndromes | |
| SS | 5 (1) |
| SC | 5 (1) |
| Carriers (heterozygous) | |
| Hemoglobin S | 285 (57) |
| Hemoglobin C | 182 (36.5) |
| Hemoglobin G-Philadelphia | 7 (1.4) |
| Hemoglobin D | 4 (0.8) |
| Hemoglobin E | 1(0.2) |
| Hemoglobin J | 1 (0.2) |
| Hemoglobin O-Arabia | 1 (0.2) |
| Others | |
| CC | 4 (0.8) |
| Alpha-thalassemia | 2 (0.4) |
| Hemoglobin C/Korle-Bu | 1 (0.2) |
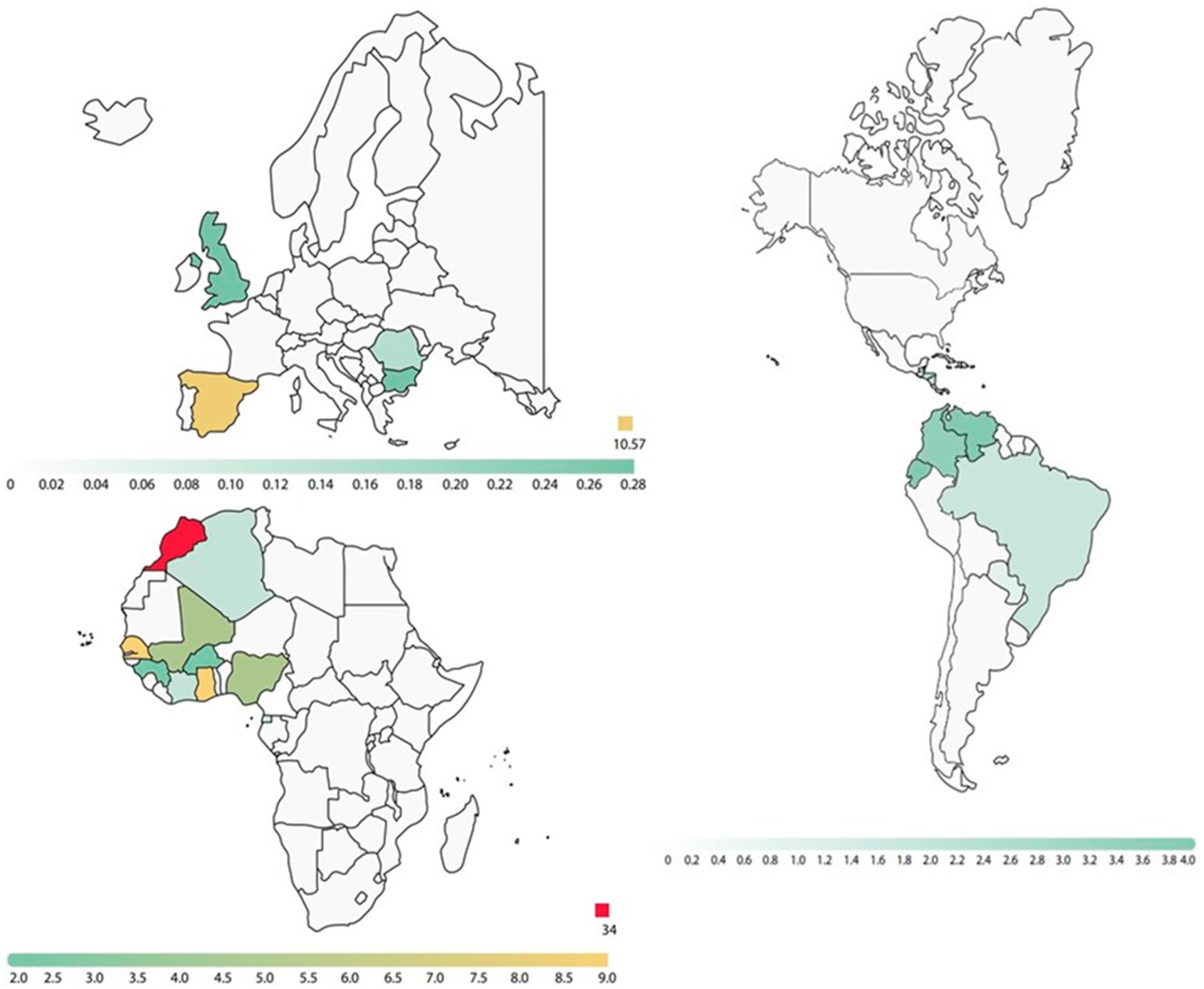
| Regions | Structural Variants Detected in Parents | ||||||
|---|---|---|---|---|---|---|---|
| AS | AC | SS | CC | SC | OTHER | ||
| Africa | Northern | 57 (6%) | 67 (7.1%) | - | 2 (0.2%) | 1 (0.1%) | 2 (0.2%) |
| Occidental | 102 (10.8%) | 49 (5.2%) | - | 6 (0.6%) | 4 (0.4%) | - | |
| Central | 4 (0.4%) | - | - | - | - | - | |
| Oriental | - | - | - | - | - | - | |
| Southern | - | 1 (0.11%) | - | - | - | - | |
| Unknown | 32 (3.4%) | 22 (2.3%) | - | 2 (0.2%) | 1 (0.1%) | 2 (0.2%) | |
| America | 57 (6%) | 20 (2.1%) | 1 (0.1%) | - | - | 1 (0.1%) | |
| Canada | 1 (0.1%) | - | - | - | - | - | |
| Europe | 9 (1%) | 4 (0.4%) | - | - | - | 6 (0.6%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Villalobos, M.; Campos Baños, E.; Juan Fita, M.J.; Egea Mellado, J.M.; Gonzalez Gallego, I.; Beltrán Videla, A.; Berenguer Piqueras, M.; Bermúdez Cortés, M.; Moraleda Jiménez, J.M.; Guillen Navarro, E.; et al. A Newborn Screening Program for Sickle Cell Disease in Murcia (Spain). Int. J. Neonatal Screen. 2023, 9, 55. https://doi.org/10.3390/ijns9040055
Sánchez-Villalobos M, Campos Baños E, Juan Fita MJ, Egea Mellado JM, Gonzalez Gallego I, Beltrán Videla A, Berenguer Piqueras M, Bermúdez Cortés M, Moraleda Jiménez JM, Guillen Navarro E, et al. A Newborn Screening Program for Sickle Cell Disease in Murcia (Spain). International Journal of Neonatal Screening. 2023; 9(4):55. https://doi.org/10.3390/ijns9040055
Chicago/Turabian StyleSánchez-Villalobos, María, Eulalia Campos Baños, María Jesús Juan Fita, José María Egea Mellado, Inmaculada Gonzalez Gallego, Asunción Beltrán Videla, Mercedes Berenguer Piqueras, Mar Bermúdez Cortés, José María Moraleda Jiménez, Encarna Guillen Navarro, and et al. 2023. "A Newborn Screening Program for Sickle Cell Disease in Murcia (Spain)" International Journal of Neonatal Screening 9, no. 4: 55. https://doi.org/10.3390/ijns9040055
APA StyleSánchez-Villalobos, M., Campos Baños, E., Juan Fita, M. J., Egea Mellado, J. M., Gonzalez Gallego, I., Beltrán Videla, A., Berenguer Piqueras, M., Bermúdez Cortés, M., Moraleda Jiménez, J. M., Guillen Navarro, E., Salido Fierrez, E., & Pérez-Oliva, A. B. (2023). A Newborn Screening Program for Sickle Cell Disease in Murcia (Spain). International Journal of Neonatal Screening, 9(4), 55. https://doi.org/10.3390/ijns9040055