
Immunoreactive Trypsinogen in Infants Born to Women with Cystic Fibrosis Taking Elexacaftor–Tezacaftor–Ivacaftor
 ,
,
Abstract
1. Introduction
2. Materials and Methods
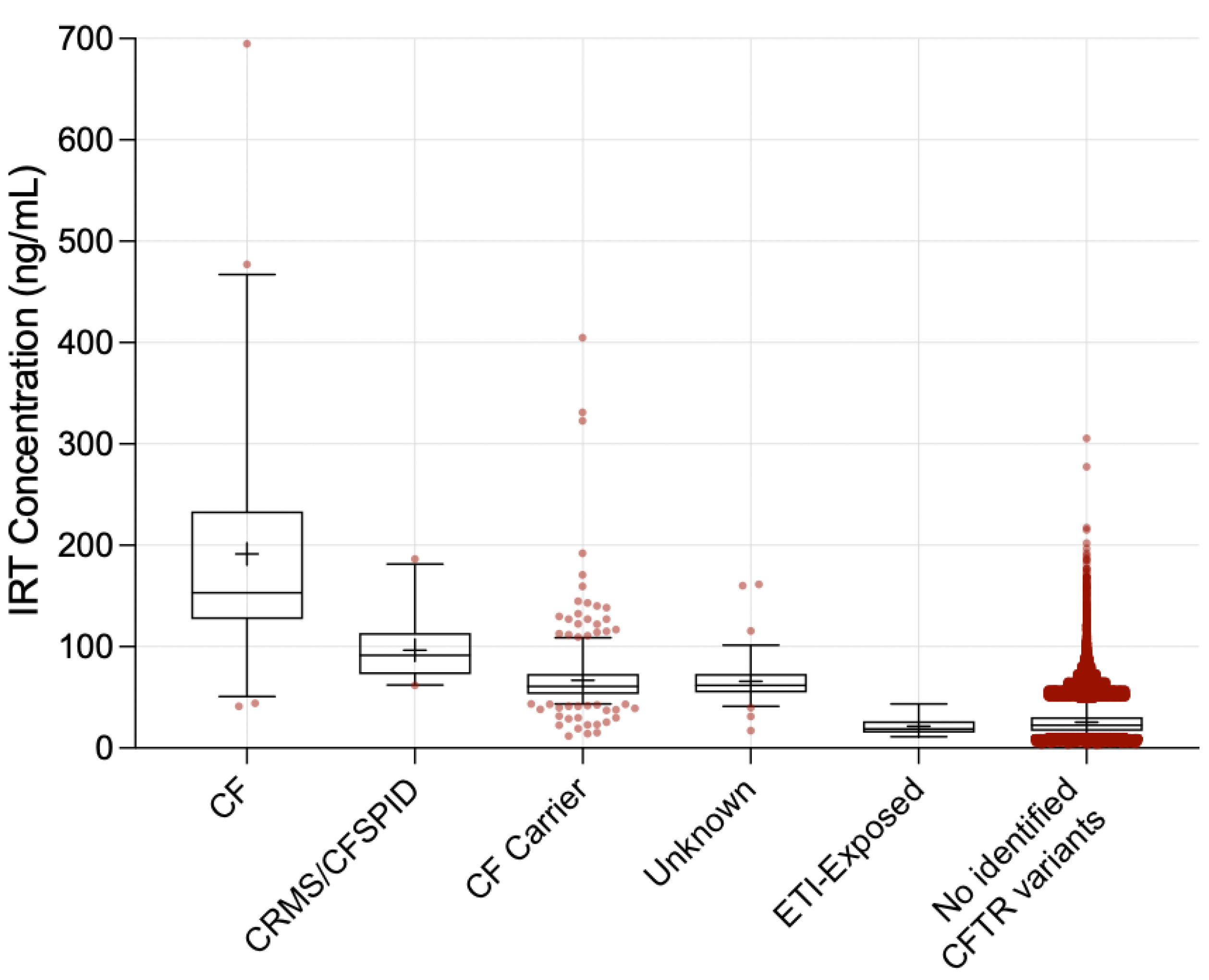
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Dwight, M.; Marshall, B. CFTR modulators: Transformative therapies for cystic fibrosis. J. Manag. Care Spec. Pharm. 2021, 27, 281–284. [Google Scholar] [CrossRef]
- Huang, Y.; Paul, G.; Lee, J.; Yarlagadda, S.; McCoy, K.; Naren, A.P. Elexacaftor/Tezacaftor/Ivacaftor Improved Clinical Outcomes in a Patient with N1303K-CFTR Based on In Vitro Experimental Evidence. Am. J. Respir. Crit. Care Med. 2021, 204, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Collins, B.; Fortner, C.; Cotey, A.; Esther, C.R.J.; Trimble, A. Drug exposure to infants born to mothers taking Elexacaftor, Tezacaftor, and Ivacaftor. J. Cyst. Fibros. 2022, 21, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yi, Y.; Yan, Z.; Rosen, B.H.; Liang, B.; Winter, M.C.; Evans, T.I.A.; Rotti, P.G.; Yang, Y.; Gray, J.S.; et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med. 2019, 11, eaau7531. [Google Scholar] [CrossRef]
- Crossley, J.R.; Elliott, R.B.; Smith, P.A. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet 1979, 1, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.R.; Aitken, M.L. Pregnancy in cystic fibrosis. Curr. Opin. Pulm. Med. 2007, 13, 537–540. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Jain, R. Maternal and fetal outcomes following elexacaftor-tezacaftor-ivacaftor use during pregnancy and lactation. J. Cyst. Fibros. 2021, 20, 402–406. [Google Scholar] [CrossRef]
- Jain, R.; Taylor-Cousar, J.L. Fertility, Pregnancy and Lactation Considerations for Women with CF in the CFTR Modulator Era. J. Pers. Med. 2021, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Fortner, C.N.; Seguin, J.M.; Kay, D.M. Normal pancreatic function and false-negative CF newborn screen in a child born to a mother taking CFTR modulator therapy during pregnancy. J. Cyst. Fibros. 2021, 20, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Dunnett, C.W. A multiple comparison procedure for comparing several treatments with a control. J. Am. Stat. Assoc. 1955, 50, 1096–1121. [Google Scholar] [CrossRef]
- Szentpetery, S.; Foil, K.; Hendrix, S.; Gray, S.; Mingora, C.; Head, B.; Johnson, D.; Flume, P.A. A case report of CFTR modulator administration via carrier mother to treat meconium ileus in a F508del homozygous fetus. J. Cyst. Fibros. 2022, 21, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Picci, L.; Scarpa, M.; Dechecchi, M.C.; Zanolla, L.; Assael, B.M.; Zacchello, F. Cystic fibrosis carriers have higher neonatal immunoreactive trypsinogen values than non-carriers. Am. J. Med. Genet. A 2005, 135, 142–144. [Google Scholar] [CrossRef]
- Rock, M.J.; Levy, H.; Zaleski, C.; Farrell, P.M. Factors accounting for a missed diagnosis of cystic fibrosis after newborn screening. Pediatr. Pulmonol. 2011, 46, 1166–1174. [Google Scholar] [CrossRef]
- Kharrazi, M.; Sacramento, C.; Comeau, A.M.; Hale, J.E.; Caggana, M.; Kay, D.M.; Lee, R.; Reilly, B.; Thompson, J.D.; Nasr, S.Z.; et al. Missed Cystic Fibrosis Newborn Screening Cases due to Immunoreactive Trypsinogen Levels below Program Cutoffs: A National Survey of Risk Factors. Int. J. Neonatal Screen. 2022, 8, 58. [Google Scholar] [CrossRef]
- Farrell, P.M.; Sommerburg, O. Toward quality improvement in cystic fibrosis newborn screening: Progress and continuing challenges. J. Cyst. Fibros. 2016, 15, 267–269. [Google Scholar] [CrossRef]
- Kay, D.M.; Maloney, B.; Hamel, R.; Pearce, M.; DeMartino, L.; McMahon, R.; McGrath, E.; Krein, L.; Vogel, B.; Saavedra-Matiz, C.A.; et al. Screening for cystic fibrosis in New York State: Considerations for algorithm improvements. Eur. J. Pediatr. 2016, 175, 181–193. [Google Scholar] [CrossRef]
- McGarry, M.E.; Ren, C.L.; Wu, R.; Farrell, P.M.; McColley, S.A. Detection of disease-causing CFTR variants in state newborn screening programs. Pediatr. Pulmonol. 2022, 58, 465–474. [Google Scholar] [CrossRef]
- Farrell, P.M.; Langfelder-Schwind, E.; Farrell, M.H. Challenging the dogma of the healthy heterozygote: Implications for newborn screening policies and practices. Mol. Genet. Metab. 2021, 134, 8–19. [Google Scholar] [CrossRef]
- Rock, M.J.; Hoffman, G.; Laessig, R.H.; Kopish, G.J.; Litsheim, T.J.; Farrell, P.M. Newborn screening for cystic fibrosis in Wisconsin: Nine-year experience with routine trypsinogen/DNA testing. J Pediatr 2005, 147 (Suppl. 3), S73–S77. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.; Kosorok, M.; Rock, M.; Laxova, A.; Zeng, L.; Lai, H.; Hoffman, G.; Laessig, R.; Splaingard, M. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 2001, 107, 1–13. [Google Scholar] [CrossRef]
- Schrijver, I.; Pique, L.; Graham, S.; Pearl, M.; Cherry, A.; Kharrazi, M. The Spectrum of CFTR Variants in Nonwhite Cystic Fibrosis Patients: Implications for Molecular Diagnostic Testing. J. Mol. Diagn. 2016, 18, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.H.; Mooney, K.E.; Laxova, A.; Farrell, P.M. Parental Preferences about Policy Options Regarding Disclosure of Incidental Genetic Findings in Newborn Screening: Using Videos and the Internet to Educate and Obtain Input. Int. J. Neonatal Screen. 2022, 8, 54. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Infant Group | N | ≥1 F508del (%) | Mean IRT (ng/mL) | Median IRT (ng/mL) | IQR | p-Value 3 |
|---|---|---|---|---|---|---|
| CF 1 | 51 | 98.0% | 191.6 | 153.1 | 127.1–233.6 | <0.001 |
| CRMS/CFSPID 1 | 21 | 80.9% | 96.7 | 91.5 | 73.0–112.2 | <0.001 |
| CF Carrier 1 | 489 | 62.8% | 66.9 | 60.8 | 53.1–73.2 | <0.001 |
| Unknown Diagnosis 1 | 75 | 65.3% | 65.7 | 61.8 | 54.8–73.1 | <0.001 |
| ETI-Exposed | 19 | n/a 2 | 21.6 | 18.9 | 15.2–26.5 | - |
| No identified CFTR variants | 189,857 | 0.0% | 25.4 | 22.5 | 16.8–30.6 | 0.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, P.; Yeley, J.; Brown, C.; Wesson, M.; Lesko, B.G.; Slaven, J.E.; Chmiel, J.F.; Jain, R.; Sanders, D.B. Immunoreactive Trypsinogen in Infants Born to Women with Cystic Fibrosis Taking Elexacaftor–Tezacaftor–Ivacaftor. Int. J. Neonatal Screen. 2023, 9, 10. https://doi.org/10.3390/ijns9010010
Patel P, Yeley J, Brown C, Wesson M, Lesko BG, Slaven JE, Chmiel JF, Jain R, Sanders DB. Immunoreactive Trypsinogen in Infants Born to Women with Cystic Fibrosis Taking Elexacaftor–Tezacaftor–Ivacaftor. International Journal of Neonatal Screening. 2023; 9(1):10. https://doi.org/10.3390/ijns9010010
Chicago/Turabian StylePatel, Payal, Jana Yeley, Cynthia Brown, Melissa Wesson, Barbara G. Lesko, James E. Slaven, James F. Chmiel, Raksha Jain, and Don B. Sanders. 2023. "Immunoreactive Trypsinogen in Infants Born to Women with Cystic Fibrosis Taking Elexacaftor–Tezacaftor–Ivacaftor" International Journal of Neonatal Screening 9, no. 1: 10. https://doi.org/10.3390/ijns9010010
APA StylePatel, P., Yeley, J., Brown, C., Wesson, M., Lesko, B. G., Slaven, J. E., Chmiel, J. F., Jain, R., & Sanders, D. B. (2023). Immunoreactive Trypsinogen in Infants Born to Women with Cystic Fibrosis Taking Elexacaftor–Tezacaftor–Ivacaftor. International Journal of Neonatal Screening, 9(1), 10. https://doi.org/10.3390/ijns9010010