
Impact of Pancreatitis-Associated Protein on Newborn Screening Outcomes and Detection of CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A Monocentric Prospective Pilot Experience



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and NBS Protocols
2.2. Statistics
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [Green Version]
- Castellani, C.; Southern, K.W.; Brownlee, K.; Dankert Roelse, J.; Duff, A.; Farrell, M.; Mehta, A.; Munck, A.; Pollitt, R.; Sermet-Gaudelus, I.; et al. European best practice guidelines for cystic fibrosis neonatal screening. J. Cyst. Fibros. 2009, 8, 153–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H.J.; Cheng, Y.; Farrell, P.M. The survival advantage of patients with cystic fibrosis diagnosed through neonatal screening: Evidence from the United States Cystic Fibrosis Foundation registry data. J. Pediatr. 2005, 147 (Suppl. S3), S57–S63. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; Kosorok, M.R.; Laxova, A.; Shen, G.; Koscik, R.E.; Bruns, W.T.; Splaingard, M.; Mischler, E.H. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. N. Engl. J. Med. 1997, 337, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Barben, J.; Castellani, C.; Dankert-Roelse, J.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.; Munck, A.; Sands, D.; Sommerburg, O.; et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J. Cyst. Fibros. 2017, 16, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Super, M. Cystic fibrosis newborn screening and detection of carriers. Arch. Dis. Child.-Fetal Neonatal Ed. 2003, 88, F448–F449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, C.L.; Borowitz, D.S.; Gonska, T.; Howenstine, M.S.; Levy, H.; Massie, J.; Milla, C.; Munck, A.; Southern, K.W. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J. Pediatr. 2017, 181, S45–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munck, A.; Mayell, S.J.; Winters, V.; Shawcross, A.; Derichs, N.; Parad, R.; Barben, J.; Southern, K.W. ECFS Neonatal Screening Working Group. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015, 14, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barben, J.; Castellani, C.; Munck, A.; Davies, J.C.; de Winter-de Groot, K.M.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.J.; McColley, S.; et al. Updated guidance on the management of children with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). J. Cyst. Fibros. 2021, 20, 810–819. [Google Scholar] [CrossRef]
- Munck, A.; Bourmaud, A.; Bellon, G.; Picq, P.; Farrell, P.M.; Study Group DPAM. Phenotype of children with inconclusive cystic fibrosis diagnosis after newborn screening. Pediatr. Pulmonol. 2020, 55, 918–928. [Google Scholar] [CrossRef]
- Terlizzi, V.; Mergni, G.; Centrone, C.; Festini, F.; Taccetti, G. Trend of sweat chloride values in a cohort of patients carrying CFTR mutations of varying clinical consequence: Is there a risk of increasing sweat chloride over time? Pediatr. Pulmonol. 2020, 55, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Claut, L.; Tosco, A.; Colombo, C.; Raia, V.; Fabrizzi, B.; Lucarelli, M.; Angeloni, A.; Cimino, G.; Castaldo, A.; et al. A survey of the prevalence, management and outcome of infants with an inconclusive diagnosis following newborn bloodspot screening for cystic fibrosis (CRMS/CFSPID) in six Italian centres. J. Cyst. Fibros. 2021, 20, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Sarles, J.; Berthézène, P.; Le Louarn, C.; Somma, C.; Perini, J.M.; Catheline, M.; Mirallié, S.; Luzet, K.; Roussey, M.; Farriaux, J.P.; et al. Combining immunoreactive trypsinogen and pancreatitis-associated protein assays, a method of newborn screening for cystic fibrosis that avoids DNA analysis. J. Pediatr. 2005, 147, 302–305. [Google Scholar] [CrossRef]
- Iovanna, J.L.; Keim, V.; Nordback, I.; Montalto, G.; Camarena, J.; Letoublon, C.; Levy, P.; Berthezene, P.; Dagorn, J.C. Serum levels of pancreatitis-associated protein as indicators of the course of acute pancreatitis. Multicentric Study Group on Acute Pancreatitis. Gastroenterology 1994, 106, 728–734. [Google Scholar] [CrossRef]
- Sommerburg, O.; Hammermann, J. Pancreatitis-Associated Protein in Neonatal Screening for Cystic Fibrosis: Strengths and Weaknesses. Int. J. Neonatal Screen. 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarles, J.; Barthellemy, S.; Férec, C.; Iovanna, J.; Roussey, M.; Farriaux, J.P.; Toutain, A.; Berthelot, J.; Maurin, N.; Codet, J.P.; et al. Blood concentrations of pancreatitis associated protein in neonates: Relevance to neonatal screening for cystic fibrosis. Arch. Dis. Child. Fetal. Neonatal Ed. 1999, 80, F118–F122. [Google Scholar] [CrossRef]
- Dankert-Roelse, J.E.; Bouva, M.J.; Jakobs, B.S.; Janssens, H.M.; de Winter-de Groot, K.M.; Schönbeck, Y.; Gille, J.J.P.; Gulmans, V.A.M.; Verschoof-Puite, R.K.; Schielen, P.C.J.I.; et al. Newborn blood spot screening for cystic fibrosis with a four-step screening strategy in the Netherlands. J. Cyst. Fibros. 2019, 18, 54–63. [Google Scholar] [CrossRef]
- Marcão, A.; Barreto, C.; Pereira, L.; Vaz, L.G.; Cavaco, J.; Casimiro, A.; Félix, M.; Silva, T.R.; Barbosa, T.; Freitas, C.; et al. Cystic Fibrosis Newborn Screening in Portugal: PAP Value in Populations with Stringent Rules for Genetic Studies. Int. J. Neonatal Screen. 2018, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Zeyda, M.; Schanzer, A.; Basek, P.; Bauer, V.; Eber, E.; Ellemunter, H.; Kallinger, M.; Riedler, J.; Thir, C.; Wadlegger, F.; et al. Cystic Fibrosis Newborn Screening in Austria Using PAP and the Numeric Product of PAP and IRT Concentrations as Second-Tier Parameters. Diagnostics 2021, 11, 299. [Google Scholar] [CrossRef]
- Sommerburg, O.; Stahl, M.; Hämmerling, S.; Gramer, G.; Muckenthaler, M.U.; Okun, J.; Kohlmüller, D.; Happich, M.; Kulozik, A.E.; Mall, M.A.; et al. Final results of the southwest German pilot study on cystic fibrosis newborn screening—Evaluation of an IRT/PAP protocol with IRT-dependent safety net: Results of the Southwest German CFNBS pilot study. J. Cyst. Fibros. 2022, 21, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Teper, A.; Smithuis, F.; Rodríguez, V.; Salvaggio, O.; Maccallini, G.; Aranda, C.; Lubovich, S.; Zaragoza, S.; García-Bournissen, F. Comparison between two newborn screening strategies for cystic fibrosis in Argentina: IRT/IRT versus IRT/PAP. Pediatr. Pulmonol. 2021, 56, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Sadik, I.; Pérez de Algaba, I.; Jiménez, R.; Benito, C.; Blasco-Alonso, J.; Caro, P.; Navas-López, V.M.; Pérez-Frías, J.; Pérez, E.; Serrano, J.; et al. Initial Evaluation of Prospective and Parallel Assessments of Cystic Fibrosis Newborn Screening Protocols in Eastern Andalusia: IRT/IRT versus IRT/PAP/IRT. Int. J. Neonatal Screen. 2019, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Werbrouck, A.; Verhaeghe, N.; De Wachter, E.; Simoens, S.; Annemans, L.; Putman, K. A model-based economic evaluation of four newborn screening strategies for cystic fibrosis in Flanders, Belgium. Acta. Clin. Belg. 2020, 75, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Sarles, J.; Giorgi, R.; Berthézène, P.; Munck, A.; Cheillan, D.; Dagorn, J.C.; Roussey, M. Neonatal screening for cystic fibrosis: Comparing the performances of IRT/DNA and IRT/PAP. J. Cyst. Fibros. 2014, 13, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.M.; White, T.B.; Howenstine, M.S.; Munck, A.; Parad, R.B.; Rosenfeld, M.; Sommerburg, O.; Accurso, F.J.; Davies, J.C.; Rock, M.J.; et al. Diagnosis of Cystic Fibrosis in Screened Populations. J. Pediatr. 2017, 181, S33–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeGrys, V.A.; Yankaskas, J.R.; Quittell, L.M.; Marshall, B.C.; Mogayzel, P.J., Jr.; Cystic Fibrosis Foundation. Cystic Fibrosis Foundation. Diagnostic sweat testing: The Cystic Fibrosis Foundation guidelines. J. Pediatr. 2007, 151, 85–89. [Google Scholar] [CrossRef]
- Christiansen, A.L.; Nybo, M. Lack of harmonization in sweat testing for cystic fibrosis—A national survey. Scand. J. Clin. Lab. Investig. 2014, 74, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Mergni, G.; Buzzetti, R.; Centrone, C.; Zavataro, L.; Braggion, C. Cystic fibrosis screen positive inconclusive diagnosis (CFSPID): Experience in Tuscany, Italy. J. Cyst. Fibros. 2019, 18, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, A.; Cimbalo, C.; Castaldo, R.J.; D’Antonio, M.; Scorza, M.; Salvadori, L.; Sepe, A.; Raia, V.; Tosco, A. Cystic Fibrosis-Screening Positive Inconclusive Diagnosis: Newborn Screening and Long-Term Follow-Up Permits to Early Identify Patients with CFTR-Related Disorders. Diagnostics 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, V.; Seia, M.; Raimondi, S.; Costantino, L.; Capasso, P.; Porcaro, L.; Porcaro, C.; Colombo, C.; Coviello, D.A.; Mariani, T.; et al. Cystic fibrosis newborn screening: Distribution of blood immunoreactive trypsinogen concentrations in hypertrypsinemic neonates. JIMD Rep. 2012, 4, 17–23. [Google Scholar]
- Thauvin-Robinet, C.; Munck, A.; Huet, F.; Génin, E.; Bellis, G.; Gautier, E.; Audrézet, M.P.; Férec, C.; Lalau, G.; Georges, M.D.; et al. The very low penetrance of cystic fibrosis for the R117H mutation: A reappraisal for genetic counselling and newborn screening. J. Med. Genet. 2009, 46, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Krulišová, V.; Balaščaková, M.; Skalická, V.; Piskáčková, T.; Holubová, A.; Paděrová, J.; Křenková, P.; Dvořáková, L.; Zemková, D.; Kračmar, P.; et al. Prospective and parallel assessments of cystic fibrosis newborn screening protocols in the Czech Republic: IRT/DNA/IRT versus IRT/PAP and IRT/PAP/DNA. Eur. J. Pediatr. 2012, 171, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Krulisova, V.; Hammermann, J.; Lindner, M.; Stahl, M.; Muckenthaler, M.; Kohlmueller, D.; Happich, M.; Kulozik, A.E.; Votava, F.; et al. Comparison of different IRT-PAP protocols to screen newborns for cystic fibrosis in three central European populations. J. Cyst. Fibros. 2014, 13, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terlizzi, V.; Claut, L.; Colombo, C.; Tosco, A.; Castaldo, A.; Fabrizzi, B.; Lucarelli, M.; Cimino, G.; Carducci, C.; Dolce, D.; et al. of early repeat sweat testing in infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/CF screen-positive, inconclusive diagnosis. Pediatr. Pulmonol. 2021, 56, 3785–3791. [Google Scholar] [CrossRef]
- Tosco, A.; Castaldo, A.; Colombo, C.; Claut, L.; Carnovale, V.; Iacotucci, P.; Lucarelli, M.; Cimino, G.; Fabrizzi, B.; Caporelli, N.; et al. Clinical outcomes of a large cohort of individuals with the F508del/5T;TG12 CFTR genotype. J. Cyst. Fibros. 2022, in press. [Google Scholar] [CrossRef]
- Terlizzi, V.; Di Lullo, A.M.; Comegna, M.; Centrone, C.; Pelo, E.; Castaldo, G.; Raia, V.; Braggion, C. S737F is a new CFTR mutation typical of patients originally from the Tuscany region in Italy. Ital. J. Pediatr. 2018, 44, 2. [Google Scholar] [CrossRef] [Green Version]
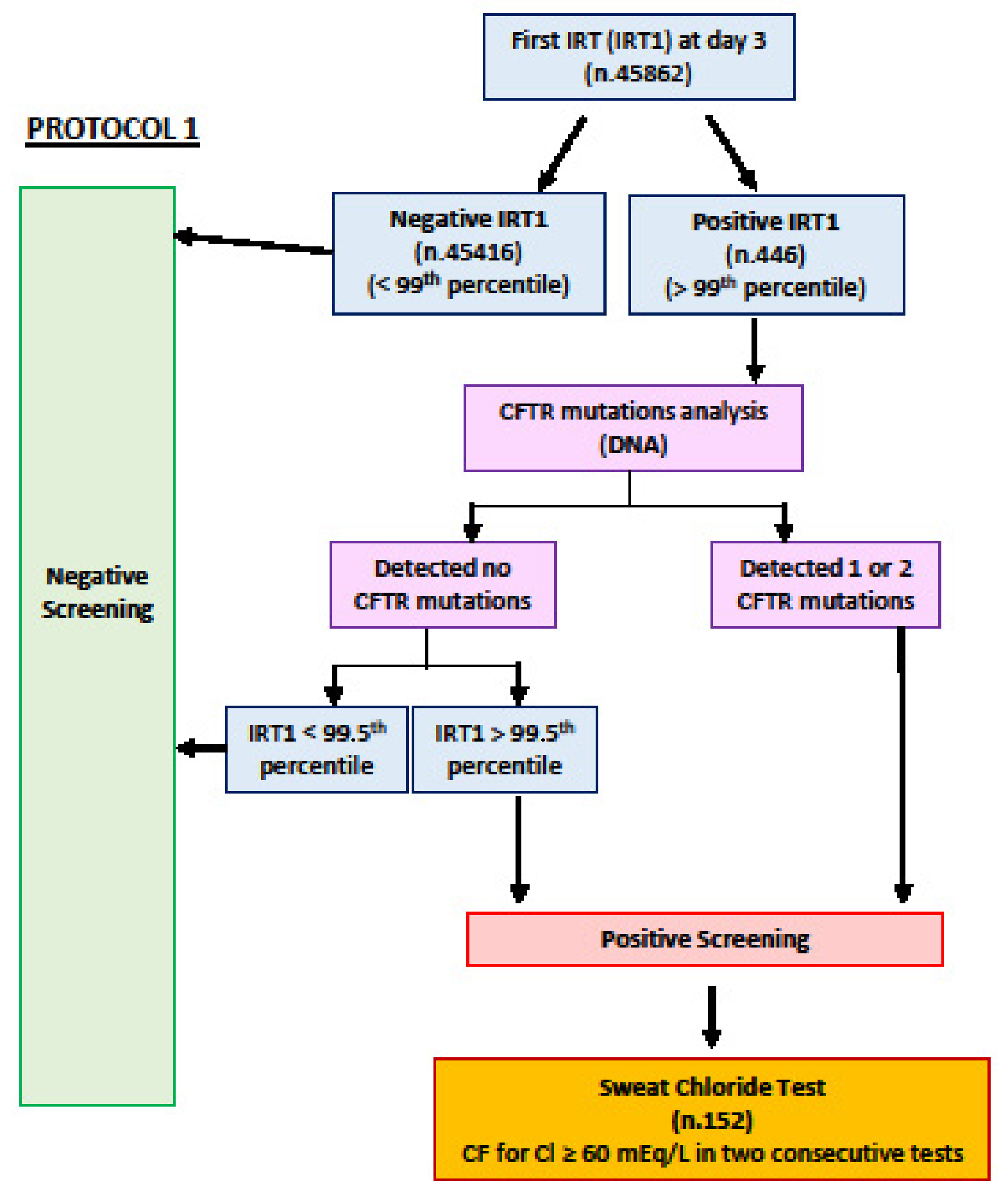

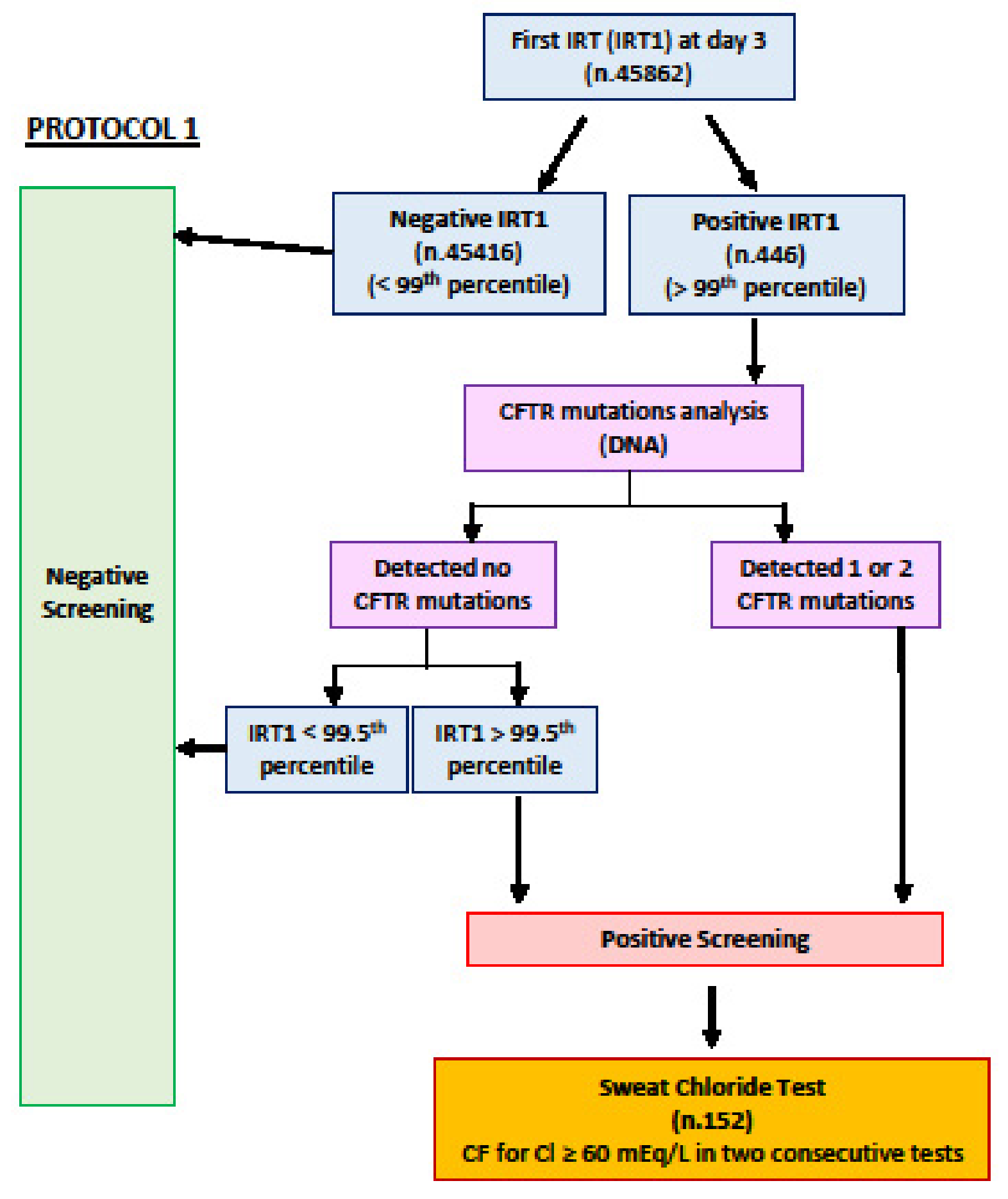{kind=link}
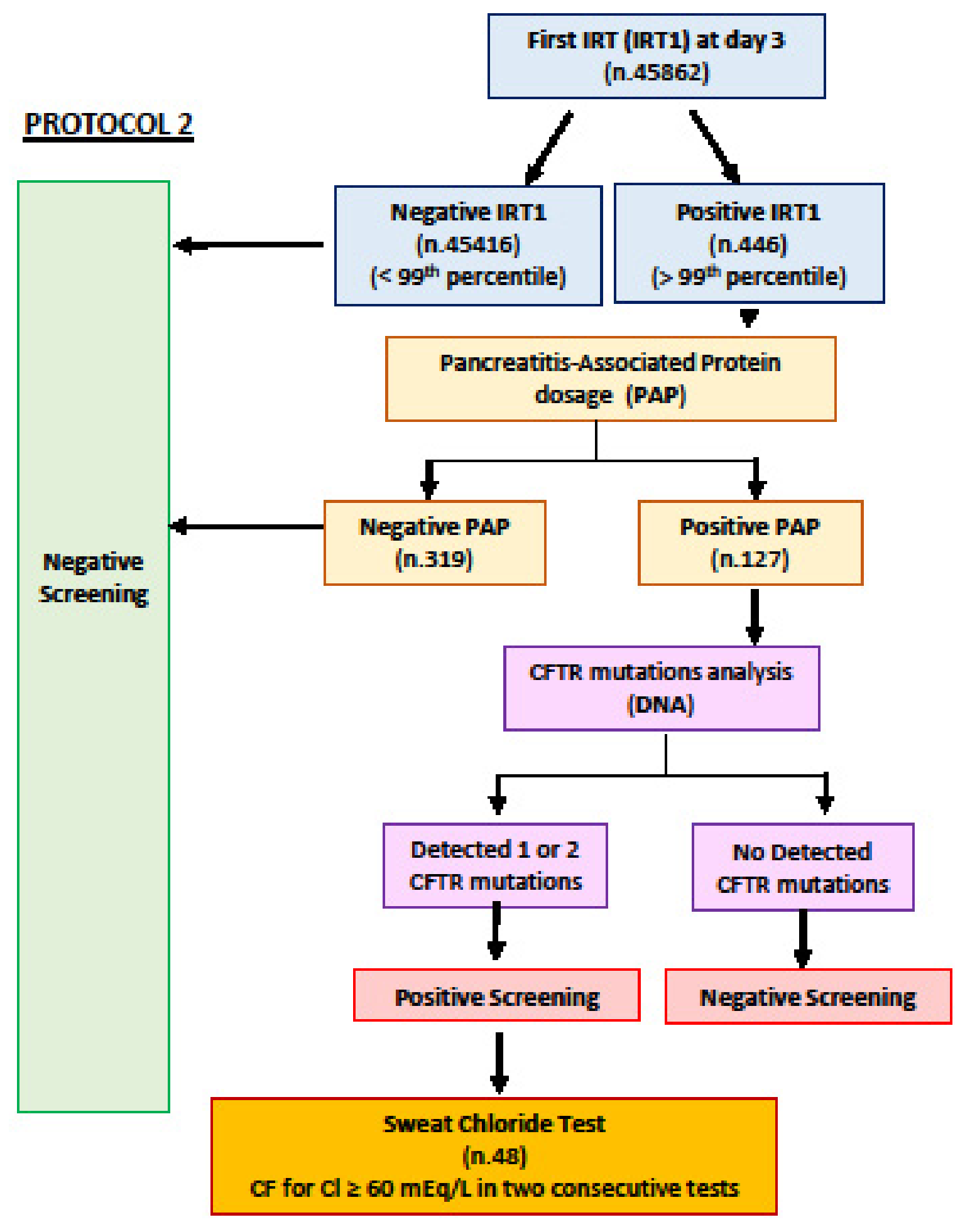
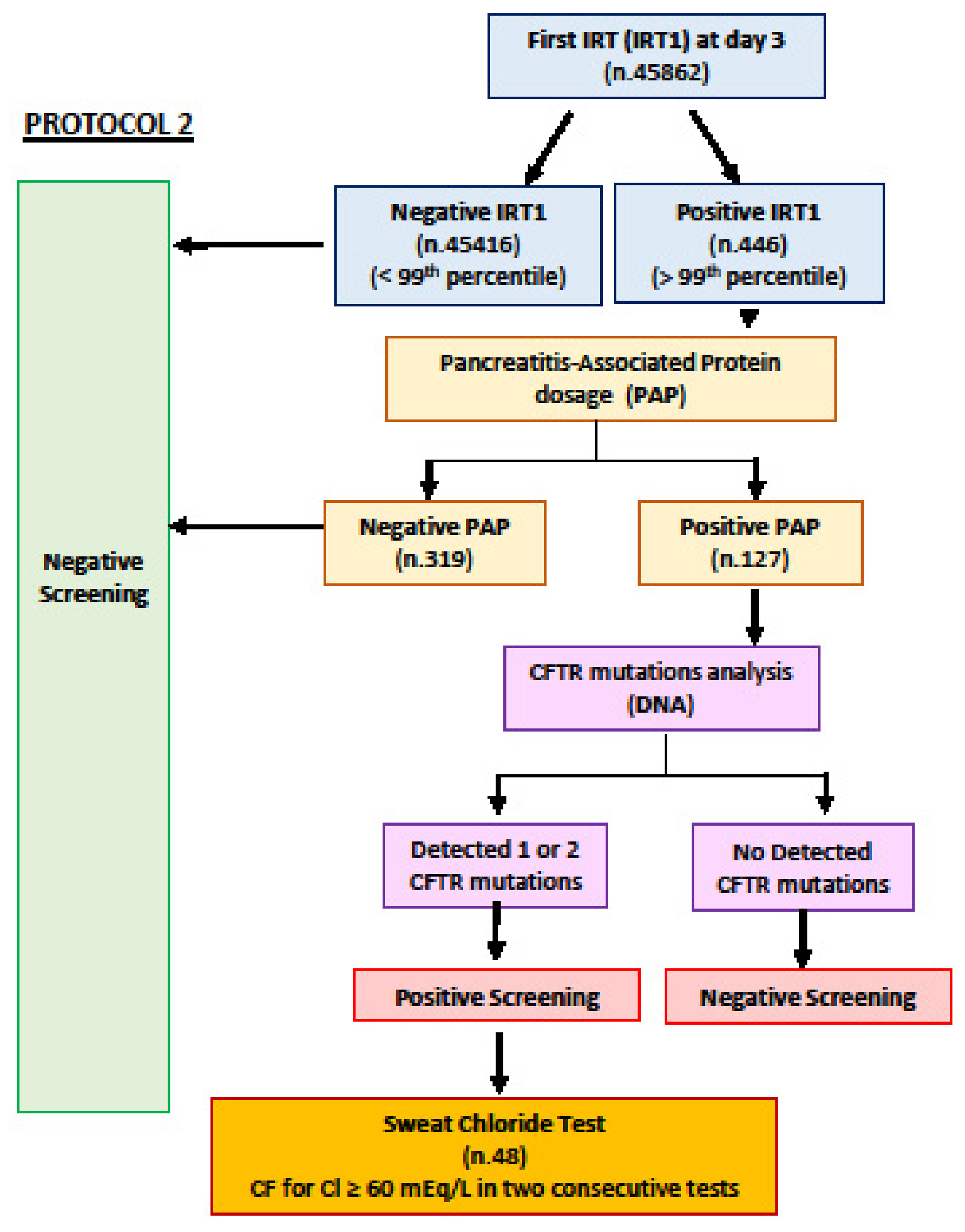{kind=link}
| IRT (ng/mL) | PAP (μg/L) | CFTR Variant 1 | CFTR Variant 2 | First SC (mEq/L) | Last SC (mEq/L), Age (Months) | Diagnosis at Study End (31 May 2022) | ||
|---|---|---|---|---|---|---|---|---|
| CF | ||||||||
| 1 | 98 | 1.53 | N1303K | N1303K | 126 | / | CF | |
| 2 | 75 | 13.95 | L867X | G378X/I148T a | 109 | / | CF | |
| 3 | 91 | 1.13 | F508del | G126D | 66 | / | CF | |
| 4 | 182 | 0.86 | F508del | F508del | 84 | / | CF | |
| 5 | 87 | 4.70 | F508del | c.870-113_870-1110delGAAT | 65 | / | CF | |
| 6 | 141 | 17.44 | F508del | F508del | 126 | / | CF | |
| 7 | 96 | 1.24 | R117C | G542X | 48 | 51 (2) | CF | |
| 8 | 71 | 2.76 | R553X | 2789+5G-> A | 93 | CF | ||
| CRMS/CFSPID | ||||||||
| 1 * | 93 | 2.07 | E585X | UN c | 49 | 16 (18) | healthy carrier | |
| 2 | 47 | 0.60 | F508del | 5T; TG12 | 50 | 45 (11) | CMRS/CFSPID | |
| 3 | 53 | 1.18 | F1052V/621+3° > G a | UN c | 32 | 18 (6) | healthy carrier | |
| 4 | 54 | 0.27 | F508del | L997F b | 36 | 29 (21) | CRMS/CFSPID | |
| 5 * | 58 | 3.01 | F508del | S912L | 36 | 48 (19) | CRMS/CFSPID | |
| 6 | 58 | 1.11 | N1303K | F508C b | 31 | 25 (19) | CRMS/CFSPID | |
| 7 * | 129 | 3.02 | 2789+5G -> A | 5T-12TG | 39 | 34 (19) | CRMS/CFSPID | |
| 8 * | 54 | 3.20 | F508del/L467P a | UN c | 31 | 31 (9) | healthy carrier | |
| 9 * | 54 | 2.01 | F508del | UN c | 49 | 18(8) | healthy carrier | |
| 10 | 65 | 1.25 | F508del | S737F | 51 | 50 (9) | CRMS/CFSPID | |
| 11 | 53 | 1.01 | L1065P | L997F b | 31 | 18 (8) | CRMS/CFSPID | |
| 12 | 60 | 0.61 | F508del | UN c | 32 | 25 (6) | healthy carrier | |
| 13 | 62 | 1.58 | D110H | M952I d | 32 | 17 (6) | CRMS/CFSPID | |
| CF vs. (CRMS/CFSPID + Healthy) | |||||
|---|---|---|---|---|---|
| IRT-DNA Protocol 1 | IRT-PAP-DNA Protocol 2 | IRT-PAP-DNA Modified Protocol 2 | p Value Protocol 1 vs. Protocol 2 | p Value Protocol 1 vs. Modified Protocol 2 | |
| Sensitivity % | 100.00 (63.06–100.00) | 100.00 (47.82–100.00) | 100.00 (63.06–100.00) | ||
| Specificity % | 99.69 (99.64–99.74) | 99.91 (99.87–99.93) | 99.90 (99.87–99.93) | <0.001 | <0.001 |
| PPV % | 5.33 (4.56–6.23) | 10.42 (7.94–13.55) | 15.38 (11.92–19.63) | 0.311 | 0.033 |
| NPV % | 100.00 | 100.00 | 100.00 | ||
| Positive LR | 322.92 (274.01–380.55) | 1066.44 (791.02–1437.76) | 1042.16 (775.66–1400.23) | <0.001 | <0.001 |
| Negative LR | 0.00 | 0.00 | 0.00 | ||
| IRT | PAP | |||
|---|---|---|---|---|
| Screening Diagnosis (Newborns Number) | Mean (ng/mL) | SD | Mean (μg/L) | SD |
| Healthy (366) | 61.17 | 38.86 | 1.86 | 5.97 |
| Healthy carriers (59) | 59.97 | 16.77 | 1.39 | 0.96 |
| CRMS/CFSPID (13) | 64.62 | 22.33 | 1.52 | 1.07 |
| CF (8) | 105.13 | 37.67 | 5.45 | 6.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchimani, C.; Dolce, D.; Centrone, C.; Campana, S.; Ravenni, N.; Orioli, T.; Camera, E.; Mergni, G.; Fevola, C.; Bonomi, P.; et al. Impact of Pancreatitis-Associated Protein on Newborn Screening Outcomes and Detection of CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A Monocentric Prospective Pilot Experience. Int. J. Neonatal Screen. 2022, 8, 46. https://doi.org/10.3390/ijns8030046
Bianchimani C, Dolce D, Centrone C, Campana S, Ravenni N, Orioli T, Camera E, Mergni G, Fevola C, Bonomi P, et al. Impact of Pancreatitis-Associated Protein on Newborn Screening Outcomes and Detection of CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A Monocentric Prospective Pilot Experience. International Journal of Neonatal Screening. 2022; 8(3):46. https://doi.org/10.3390/ijns8030046
Chicago/Turabian StyleBianchimani, Chiara, Daniela Dolce, Claudia Centrone, Silvia Campana, Novella Ravenni, Tommaso Orioli, Erica Camera, Gianfranco Mergni, Cristina Fevola, Paolo Bonomi, and et al. 2022. "Impact of Pancreatitis-Associated Protein on Newborn Screening Outcomes and Detection of CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A Monocentric Prospective Pilot Experience" International Journal of Neonatal Screening 8, no. 3: 46. https://doi.org/10.3390/ijns8030046
APA StyleBianchimani, C., Dolce, D., Centrone, C., Campana, S., Ravenni, N., Orioli, T., Camera, E., Mergni, G., Fevola, C., Bonomi, P., Taccetti, G., & Terlizzi, V. (2022). Impact of Pancreatitis-Associated Protein on Newborn Screening Outcomes and Detection of CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A Monocentric Prospective Pilot Experience. International Journal of Neonatal Screening, 8(3), 46. https://doi.org/10.3390/ijns8030046