
The Alberta Newborn Screening Approach for Sickle Cell Disease: The Advantages of Molecular Testing

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Newborn Screening Specimen Collection
2.3. Hemoglobin Pattern Analysis
2.4. SCD Screening Algorithm
2.5. Molecular Genetic Testing
2.6. Calling Out Abnormal Screening Results
3. Results
3.1. Screened Infants
3.2. Screening of Transfused Infants
3.3. Positive Screening Results for SCD
3.4. Hemoglobin Variant Carriers
3.5. False Positive Screening Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| α | Alpha |
| β | Beta |
| γ | Gamma |
| BG | Biochemical geneticist |
| BW | Birth weight |
| d | Day |
| DBS | Dried blood spot |
| FP | False positive |
| FSa | HbS/beta thalassemia |
| GA | Gestational age |
| Hb | Hemoglobin |
| HbA | Adult hemoglobin |
| HbF | Fetal hemoglobin |
| HPFH | Hereditary persistence of fetal hemoglobin |
| HPLC | High-performance liquid chromatography |
| HSCT | Hematopoietic stem cell transplantation |
| IUT | Intrauterine transfusion |
| NBS | Newborn screening |
| NMS | Newborn metabolic screening |
| PCR | Polymerase chain reaction |
| RBC | Red blood cells |
| RBCT | Red blood cell transfusion |
| SCA | Sickle cell anemia |
| SCD | Sickle cell disease |
| TP | True positive |
| w | Week |
References
- Sankaran, V.G.; Orkin, S.H. The switch from fetal to adult hemoglobin. Cold Spring Harb. Perspect. Med. 2013, 3, a011643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinjamur, D.S.; Bauer, D.E.; Orkin, S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018, 180, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Forget, B.G.; Bunn, H.F. Classification of the disorders of hemoglobin. Cold Spring Harb. Perspect. Med. 2013, 3, a011684. [Google Scholar] [CrossRef] [Green Version]
- Kohne, E. Hemoglobinopathies: Clinical manifestations, diagnosis, and treatment. Dtsch. Arztebl. Int. 2011, 108, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Habara, A.; Steinberg, M.H. Minireview: Genetic basis of heterogeneity and severity in sickle cell disease. Exp. Biol. Med. 2016, 241, 689–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, M.A. Sickle Cell Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Xu, J.Z.; Thein, S.L. The carrier state for sickle cell disease is not completely harmless. Haematologica 2019, 104, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.C.; Bonham, V.L.; Joiner, C.H.; Kato, G.J.; Noonan, A.S.; Steinberg, M.H. Framing the research agenda for sickle cell trait: Building on the current understanding of clinical events and their potential implications. Am. J. Hematol. 2012, 87, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Lilley, M.; Hoang, S.; Blumenschein, P.; Peturson, A.M.; Sosova, I.; Macneil, L.; Ridsdale, R.; Christian, S. Sickle cell trait newborn screen results: Disclosure and management. J. Community Genet. 2021, 12, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Tsaras, G.; Owusu-Ansah, A.; Boateng, F.O.; Amoateng-Adjepong, Y. Complications associated with sickle cell trait: A brief narrative review. Am. J. Med. 2009, 122, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, J.F.; Thein, S.L.; Eaton, W.A. Treating sickle cell anemia. Science 2020, 367, 1198–1199. [Google Scholar] [CrossRef]
- Eaton, W.A. Linus Pauling and sickle cell disease. Biophys. Chem. 2003, 100, 109–116. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications (accessed on 4 October 2021).
- Quinn, C.T. Sickle cell disease in childhood: From newborn screening through transition to adult medical care. Pediatr. Clin. N. Am. 2013, 60, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frommel, C. Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies: A Short Review on Classical Laboratory Methods-Isoelectric Focusing, HPLC, and Capillary Electrophoresis. Int. J. Neonatal. Screen. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Haj, N.; Hoppe, C.C. Newborn Screening for SCD in the USA and Canada. Int. J. Neonatal. Screen. 2018, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Vichinsky, E.; Hurst, D.; Earles, A.; Kleman, K.; Lubin, B. Newborn screening for sickle cell disease: Effect on mortality. Pediatrics 1988, 81, 749–755. [Google Scholar]
- De Souza, A.; Wolan, V.; Battochio, A.; Christian, S.; Hume, S.; Johner, G.; Lilley, M.; Ridsdale, R.; Schnabl, K.; Tran, C.; et al. Newborn Screening: Current Status in Alberta, Canada. Int. J. Neonatal. Screen. 2019, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaf, B.; Patin, F.; Elion, J.; Couque, N. New approach to accurate interpretation of sickle cell disease newborn screening by applying multiple of median cutoffs and ratios. Pediatr. Blood Cancer 2018, 65, e27230. [Google Scholar] [CrossRef]
- Center for Disease Control and Prevention. Hemoglobinopathies: Current Practices for Screening, Confirmation and Follow-Up. Available online: https://www.cdc.gov/ncbddd/sicklecell/documents/nbs_hemoglobinopathy-testing_122015.pdf (accessed on 4 October 2021).
- Angastiniotis, M.; Eleftheriou, A.; Galanello, R.; Harteveld, C.L.; Petrou, M.; Traeger-Synodinos, J.; Giordano, P.; Jauniaux, E.; Modell, B.; Serour, G.; et al. Newborn screening for haemoglobinopathies. In Prevention of Thalassaemias and Other Haemoglobin Disorders: Volume 1: Principles; Thalassaemia International Federation: Nicosia, Cyprus, 2013; Chapter 10. [Google Scholar]
- Bouva, M.J.; Sollaino, C.; Perseu, L.; Galanello, R.; Giordano, P.C.; Harteveld, C.L.; Cnossen, M.H.; Schielen, P.C.; Elvers, L.H.; Peters, M. Relationship between neonatal screening results by HPLC and the number of alpha-thalassaemia gene mutations; consequences for the cut-off value. J. Med. Screen. 2011, 18, 182–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombatti, R.; Cela, E.; Elion, J.; Lobitz, S. Editorial for Special Issue “Newborn Screening for Sickle Cell Disease and Other Haemoglobinopathies”. Int. J. Neonatal. Screen. 2019, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global burden of sickle cell anaemia in children under five, 2010–2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [Green Version]
- Piel, F.B.; Tatem, A.J.; Huang, Z.; Gupta, S.; Williams, T.N.; Weatherall, D.J. Global migration and the changing distribution of sickle haemoglobin: A quantitative study of temporal trends between 1960 and 2000. Lancet Glob. Health 2014, 2, e80–e89. [Google Scholar] [CrossRef] [Green Version]
- Ballardini, E.; Tarocco, A.; Marsella, M.; Bernardoni, R.; Carandina, G.; Melandri, C.; Guerra, G.; Patella, A.; Zucchelli, M.; Ferlini, A.; et al. Universal neonatal screening for sickle cell disease and other haemoglobinopathies in Ferrara, Italy. Blood Transfus. 2013, 11, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L., Jr.; Lloyd-Puryear, M.A.; Eckman, J.R.; Mann, M.Y. Newborn screening for sickle cell diseases in the United States: A review of data spanning 2 decades. Semin. Perinatol. 2015, 39, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, N.; Delvin, E.E.; Hume, H.A. Newborn screening for sickle cell disease: A 1988-2003 Quebec experience. Paediatr. Child Health 2006, 11, 223–227. [Google Scholar] [CrossRef]
- Orphanet Version 5.45.0. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=232 (accessed on 4 October 2021).
- Monagel, D.; Leaker, M.; Le, D.; Wright, N.A.; Ruzycki, A.; Steele, M. Lack of an Alberta Newborn Screening Program Is Associated with Delayed Diagnosis of Sickle Cell Disease. Blood 2017, 130, 2255. [Google Scholar]
- Nusrat, M.; Moiz, B.; Nasir, A.; Rasool Hashmi, M. An insight into the suspected HbA2′ cases detected by high performance liquid chromatography in Pakistan. BMC Res. Notes 2011, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, J.D.; Scheidt, R.M. Identification of hemoglobin variants by HPLC. Clin. Chem. 2005, 51, 1303–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustace, T.; Fleisher, J.M.; Sanchez Varela, A.M.; Podda, A.; Alvarez, O. Increased prevalence of false positive hemoglobinopathy newborn screening in premature infants. Pediatr. Blood Cancer 2011, 57, 1039–1043. [Google Scholar] [CrossRef]
- Piel, F.B.; Adamkiewicz, T.V.; Amendah, D.; Williams, T.N.; Gupta, S.; Grosse, S.D. Observed and expected frequencies of structural hemoglobin variants in newborn screening surveys in Africa and the Middle East: Deviations from Hardy-Weinberg equilibrium. Genet. Med. 2016, 18, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Reed, W.; Lane, P.A.; Lorey, F.; Bojanowski, J.; Glass, M.; Louie, R.R.; Lubin, B.H.; Vichinsky, E.P. Sickle-cell disease not identified by newborn screening because of prior transfusion. J. Pediatr. 2000, 136, 248–250. [Google Scholar] [CrossRef]
- Streetly, A.; Sisodia, R.; Dick, M.; Latinovic, R.; Hounsell, K.; Dormandy, E. Evaluation of newborn sickle cell screening programme in England: 2010–2016. Arch. Dis. Child 2018, 103, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NHS. Sickle Cell and Thalassaemia Screening Programme, Handbook for Laboratories, 4th ed.; Public Health England: London, UK, 2017.
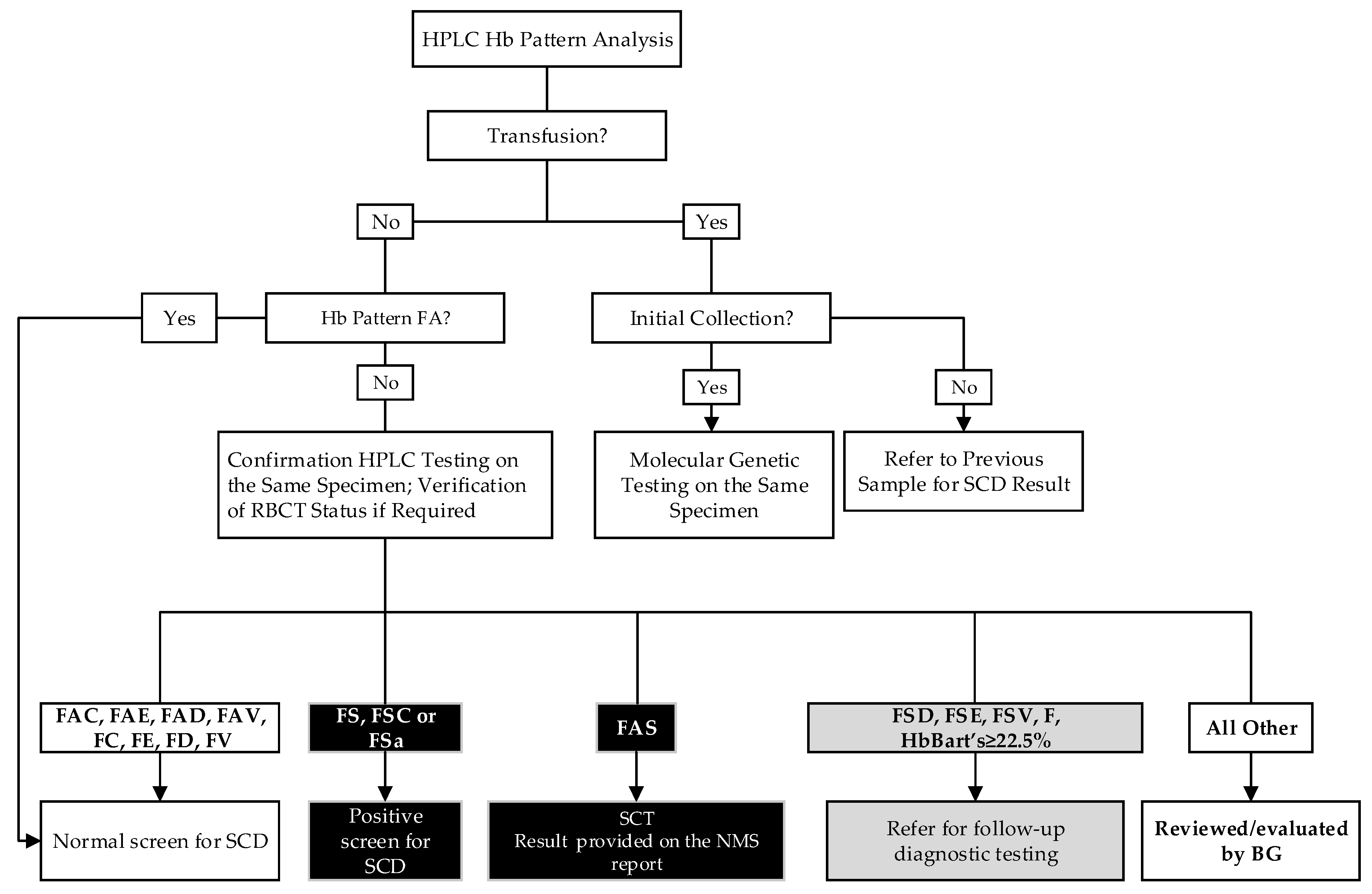
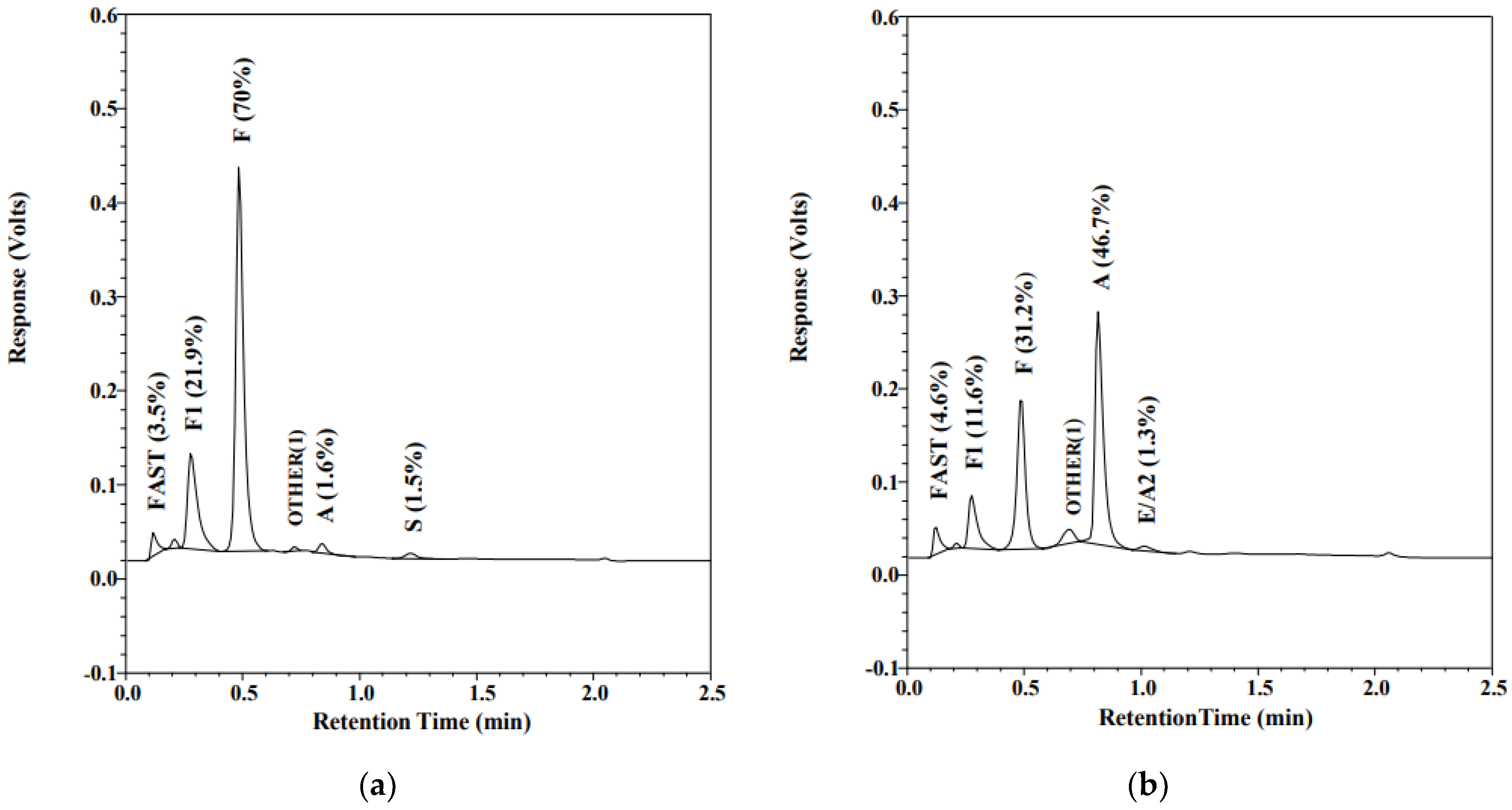

{kind=link}
{kind=link}
| Screened Infants | n | RBCT |
|---|---|---|
| Full-term | 74,343 | 102 |
| Preterm | 5971 | 86 |
| Late Preterm 34 to <37 w | 4416 | 24 |
| Moderate Preterm 32 to <34 w | 751 | 11 |
| Very preterm 28 to <32 w | 543 | 16 |
| Extremely Preterm <28 w | 261 | 35 |
| Total | 80,314 | 188 * |
| Condition * | n | Rate per 10,000 Newborns Screened |
|---|---|---|
| HbSS | 24 | 3.0 |
| HbSC | 9 | 1.1 |
| HbS/β thalassemia | 1 | 0.1 |
| HbS/HPFH | 1 | 0.1 |
| HbEE | 1 | 0.1 |
| β thalassemia major | 2 | 0.2 |
| Carriers of HbS (FAS) | 608 | 75.7 |
| Carriers of HbE (FAE) | 227 | 28.3 |
| Carriers of HbC (FAC) | 197 | 24.5 |
| Carriers of HbD (FAD) | 161 | 20.0 |
| Carrier of HbQ-Iran | 1 | 0.1 |
| Carrier of HbV (FAV) | 1 | 0.1 |
| Infant | NBS Result * | Gender | GA | Transfusion | Confirmatory Testing Result |
|---|---|---|---|---|---|
| FP1 | FAS | M | 36 w | IUT | Carrier of HbS (FAS) |
| FP2 | FSA5 | M | FT | No | Alpha-2 globin gene variant (HbQ-Iran) |
| FP3 | FSA5 | M | FT | No | Unknown alpha chain variant |
| FP4 | FA | F | FT | No | Unknown beta chain variant |
| FP5 | FAS | F | FT | No | Carrier of HbS (FAS) |
| FP6 | FSA | M | FT | No | Carrier of HbS (FAS) |
| FP7 | FSA | F | FT | No | Carrier of HbS (FAS) |
| FP8 | FSA | M | FT | No | Carrier of HbS (FAS) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.R.; Ridsdale, R.; MacNeil, L.; Lilley, M.; Hoang, S.; Christian, S.; Blumenschein, P.; Wolan, V.; Bruce, A.; Singh, G.; et al. The Alberta Newborn Screening Approach for Sickle Cell Disease: The Advantages of Molecular Testing. Int. J. Neonatal Screen. 2021, 7, 78. https://doi.org/10.3390/ijns7040078
Zhou JR, Ridsdale R, MacNeil L, Lilley M, Hoang S, Christian S, Blumenschein P, Wolan V, Bruce A, Singh G, et al. The Alberta Newborn Screening Approach for Sickle Cell Disease: The Advantages of Molecular Testing. International Journal of Neonatal Screening. 2021; 7(4):78. https://doi.org/10.3390/ijns7040078
Chicago/Turabian StyleZhou, Janet R., Ross Ridsdale, Lauren MacNeil, Margaret Lilley, Stephanie Hoang, Susan Christian, Pamela Blumenschein, Vanessa Wolan, Aisha Bruce, Gurpreet Singh, and et al. 2021. "The Alberta Newborn Screening Approach for Sickle Cell Disease: The Advantages of Molecular Testing" International Journal of Neonatal Screening 7, no. 4: 78. https://doi.org/10.3390/ijns7040078
APA StyleZhou, J. R., Ridsdale, R., MacNeil, L., Lilley, M., Hoang, S., Christian, S., Blumenschein, P., Wolan, V., Bruce, A., Singh, G., Wright, N., Parboosingh, J. S., Lamont, R. E., & Sosova, I. (2021). The Alberta Newborn Screening Approach for Sickle Cell Disease: The Advantages of Molecular Testing. International Journal of Neonatal Screening, 7(4), 78. https://doi.org/10.3390/ijns7040078