
Early Diagnosis of Classic Homocystinuria in Kuwait through Newborn Screening: A 6-Year Experience


 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. NBS Registry
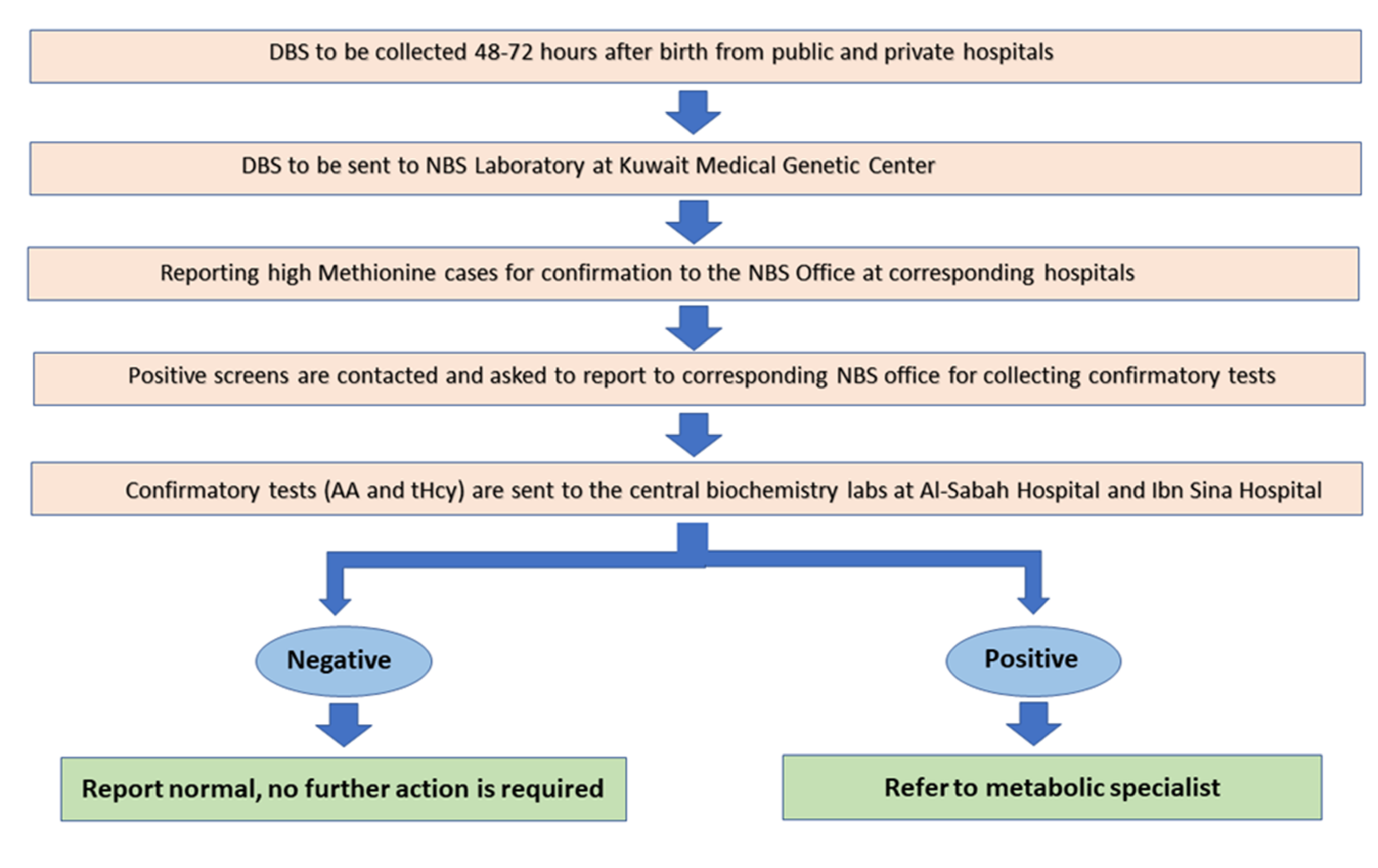
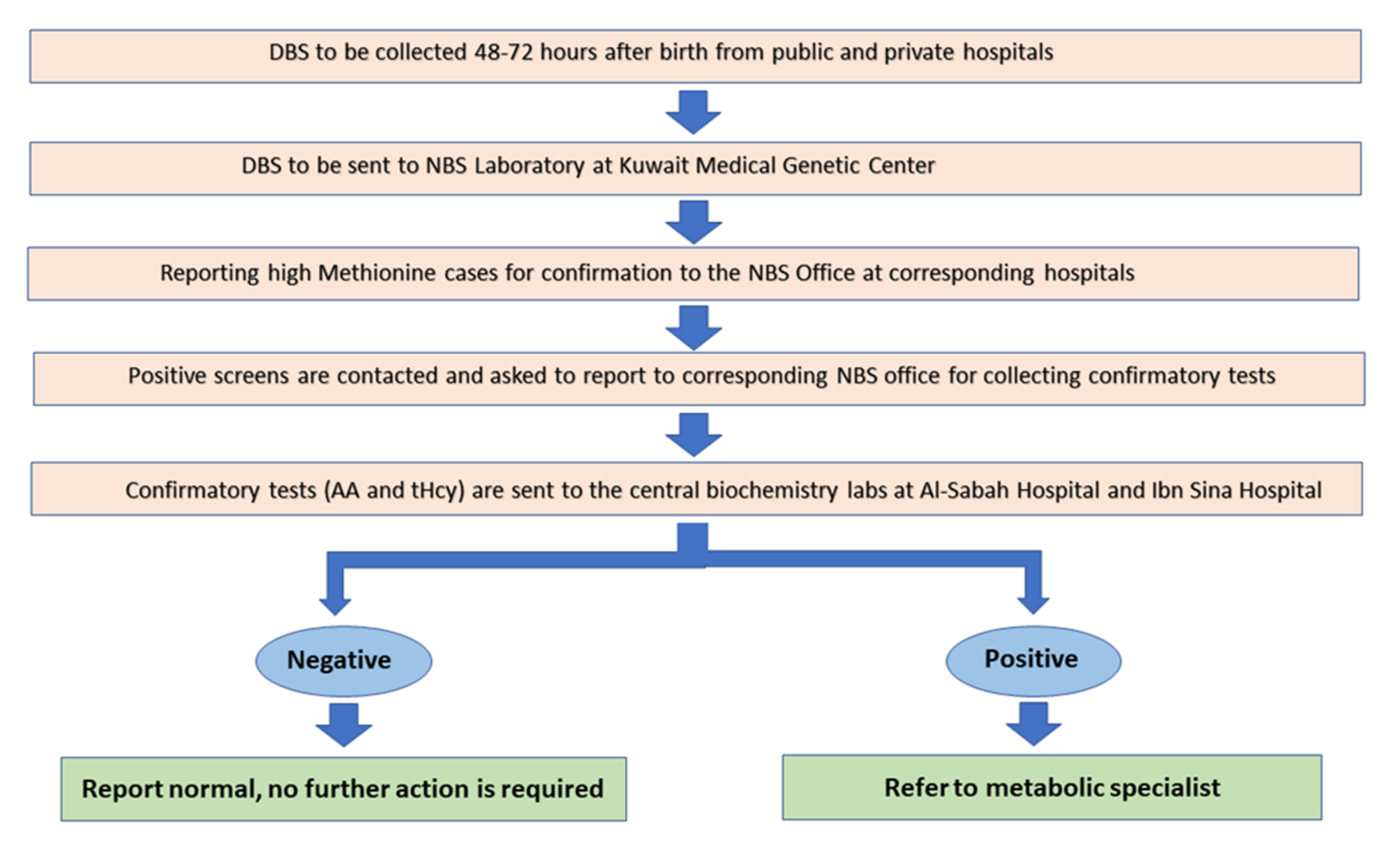
2.2. DBS Collection Protocol
2.3. Analytical Methods
2.3.1. First DBS
2.3.2. Amino Acids in Repeat (Second) DBS:
2.3.3. Plasma Amino Acids
2.3.4. Plasma Total Homocysteine
2.3.5. Evaluation of Met/Phe Ratio
2.4. Molecular Testing
3. Results
3.1. NBS Registry
3.2. DBS with Hypermethioninemia
3.3. Evaluation of Met/Phe Ratio as a Potential Strategy in Screening for HCU in Kuwait
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kuwait Government Online Geography of Kuwait. Available online: https://www.e.gov.kw/sites/kgoenglish/Pages/Visitors/AboutKuwait/KuwaitAtaGlaneGeographicalLocation.aspx (accessed on 14 April 2021).
- Kuwait Government Online Citizens and Residents. Available online: https://www.e.gov.kw/sites/kgoenglish/Pages/CitizensResidents/citizensAndResidents.aspx (accessed on 14 April 2021).
- Al-Awadi, S.A.; Moussa, M.A.; Naghuib, K.K.; Farag, T.I.; Teebi, A.S.; El-Khalifa, M.; El-Dossary, L. Consanguinity among the Kuwaiti population. Clin. Genet. 2008, 27, 483–486. [Google Scholar] [CrossRef]
- Tadmouri, G.O.; Nair, P.; Obeid, T.; Al Ali, M.T.; Al Khaja, N.; Hamamy, H.A. Consanguinity and reproductive health among Arabs. Reprod. Health 2009, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Hoss, G.R.W.; Sperb-Ludwig, F.; Schwartz, I.V.D.; Blom, H.J. Classical homocystinuria: A common inborn error of metabolism? An epidemiological study based on genetic databases. Mol. Genet. Genom. Med. 2020, 8, e1214. [Google Scholar] [CrossRef] [Green Version]
- El-Said, M.F.; Badii, R.; Bessisso, M.; Shahbek, N.; El-Ali, M.G.; El-Marikhie, M.; El-Zyoid, M.; Salem, M.; Bener, A.; Hoffmann, G.F.; et al. A common mutation in theCBSgene explains a high incidence of homocystinuria in the Qatari population. Hum. Mutat. 2006, 27, 719. [Google Scholar] [CrossRef]
- Gan-Schreier, H.; Kebbewar, M.; Fang-Hoffmann, J.; Wilrich, J.; Abdoh, G.; Ben-Omran, T.; Shahbek, N.; Bener, A.; Al Rifai, H.; Al Khal, A.L.; et al. Newborn Population Screening for Classic Homocystinuria by Determination of Total Homocysteine from Guthrie Cards. J. Pediatr. 2010, 156, 427–432. [Google Scholar] [CrossRef]
- Ismail, H.M.; Krishnamoorthy, N.; Al-Dewik, N.; Zayed, H.; Mohamed, N.A.; Di Giacomo, V.; Gupta, S.; Häberle, J.; Thöny, B.; Blom, H.J.; et al. In silico and in vivo models for Qatari-specific classical homocystinuria as basis for development of novel therapies. Hum. Mutat. 2019, 40, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Fredriksen, Å.; Meyer, K.; Ueland, P.M.; Kase, B.F. Birth prevalence of homocystinuria. J. Pediatr. 2004, 144, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Skovby, F.; Gaustadnes, M.; Mudd, S.H. A revisit to the natural history of homocystinuria due to cystathionine β-synthase deficiency. Mol. Genet. Metab. 2010, 99, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H.; Finkelstein, J.D.; Irreverre, F.; Laster, L. Homocystinuria: An Enzymatic Defect. Science 1964, 143, 1443–1445. [Google Scholar] [CrossRef]
- Mudd, S.H.; Skovby, F.; Levy, H.L. The natural history of homocystinura due to cystathionine β-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar]
- Morris, A.A.M.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.M.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.; Chrastina, P.; Pavlikova, M.; Gouveia, S.; Ribes, A.; Kölker, S.; Blom, H.J.; Baumgartner, M.R.; Bártl, J.; Dionisi-Vici, C.; et al. Newborn screening for homocystinurias: Recent recommendations versus current practice. J. Inherit. Metab. Dis. 2019, 42, 128–139. [Google Scholar] [CrossRef]
- Huemer, M.; Kožich, V.; Rinaldo, P.; Baumgartner, M.R.; Merinero, B.; Pasquini, E.; Ribes, A.; Blom, H. Newborn screening for homocystinurias and methylation disorders: Systematic review and proposed guidelines. J. Inherit. Metab. Dis. 2015, 38, 1007–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sadeq, D.W.; Nasrallah, G.K. The Spectrum of Mutations of Homocystinuria in the MENA Region. Genes 2020, 11, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ACT Sheets and Algorithms. Available online: https://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/ACT_Sheets_and_Algorithms.aspx (accessed on 13 April 2021).
- Wilcken, B.; Wiley, V.; Hammond, J.; Carpenter, K. Screening Newborns for Inborn Errors of Metabolism by Tandem Mass Spectrometry. N. Engl. J. Med. 2003, 348, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Lindner, M.; Kohlmüller, D.; Olgemöller, K.; Mayatepek, E.; Hoffmann, G.F. Expanded Newborn Screening for Inborn Errors of Metabolism by Electrospray Ionization-Tandem Mass Spectrometry: Results, Outcome, and Implications. Pediatrics 2003, 111, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Rashed, M.S.; Ozand, P.T.; Bucknall, M.; Little, D. Diagnosis of Inborn Errors of Metabolism from Blood Spots by Acylcarnitines and Amino Acids Profiling Using Automated Electrospray Tandem Mass Spectrometry. Pediatr. Res. 1995, 38, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Schulze, A.; Kohlmueller, D.; Mayatepek, E. Sensitivity of electrospray-tandem mass spectrometry using the phenylalanine/tyrosine-ratio for differential diagnosis of hyperphenylalaninemia in neonates. Clin. Chim. Acta 1999, 283, 15–20. [Google Scholar] [CrossRef]
- Pei, J.; Li, X.-Y. Determination of underivatized amino acids by high-performance liquid chromatography and electrochemical detection at an amino acid oxidase immobilized CuPtCl 6 modified electrode. Anal. Bioanal. Chem. 2000, 367, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Shipchandler, M.T.; Moore, E.G. Rapid, fully automated measurement of plasma homocyst(e)ine with the Abbott IMx analyzer. Clin. Chem. 1995, 41, 991–994. [Google Scholar] [CrossRef]
- Nexo, E.; Engbaek, F.; Ueland, P.M.; Westby, C.; O’Gorman, P.; Johnston, C.; Kase, B.F.; Guttormsen, A.B.; Alfheim, I.; McPartlin, J.; et al. Evaluation of Novel Assays in Clinical Chemistry: Quantification of Plasma Total Homocysteine. Clin. Chem. 2000, 46, 1150–1156. [Google Scholar] [CrossRef]
- Bártl, J.; Chrastina, P.; Krijt, J.; Hodík, J.; Pešková, K.; Kožich, V. Simultaneous determination of cystathionine, total homocysteine, and methionine in dried blood spots by liquid chromatography/tandem mass spectrometry and its utility for the management of patients with homocystinuria. Clin. Chim. Acta 2014, 437, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P.; Korson, M.; Jethva, R.; Allen, R.H.; Kraus, J.P.; Spector, E.B.; Wagner, C.; Mudd, S.H. Metabolic profiling of total homocysteine and related compounds in hyperho-mocysteinemia: Utility and limitations in diagnosing the cause of puzzling thrombophilia in a family. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; pp. 149–163. [Google Scholar]
- Mudd, S.H. Hypermethioninemias of genetic and non-genetic origin: A review. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Chace, D.H.; Hillman, S.L.; Millington, D.S.; Kahler, S.G.; Adam, B.W.; Levy, H.L. Rapid diagnosis of homocystinuria and other hypermethioninemias from newborns’ blood spots by tandem mass spectrometry. Clin. Chem. 1996, 42, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okun, J.G.; Gan-Schreier, H.; Ben-Omran, T.; Schmidt, K.V.; Fang-Hoffmann, J.; Gramer, G.; Abdoh, G.; Shahbeck, N.; Al Rifai, H.; Al Khal, A.L.; et al. Newborn Screening for Vitamin B6 Non-responsive Classical Homo-cystinuria: Systematical Evaluation of a Two-Tier Strategy. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2016; pp. 87–94. [Google Scholar]
- Hoedt, A.E.T.; Van Kempen, A.; Boelen, A.; Duran, M.; Kemper-Proper, E.A.; Oey-Spauwen, M.J.W.; Wijburg, F.A.; Bosch, A.M. High incidence of hypermethioninaemia in a single neonatal intensive care unit detected by a newly introduced neonatal screening programme. J. Inherit. Metab. Dis. 2007, 30, 978. [Google Scholar] [CrossRef]
- Peterschmitt, M.J.; Simmons, J.R.; Levy, H.L. Reduction of False Negati.ve Results in Screening of Newborns for Homocystinuria. N. Engl. J. Med. 1999, 341, 1572–1576. [Google Scholar] [CrossRef]
- Turgeon, C.T.; Magera, M.J.; Cuthbert, C.D.; Loken, P.R.; Gavrilov, D.K.; Tortorelli, S.; Raymond, K.M.; Oglesbee, D.; Rinaldo, P.; Matern, D. Determination of Total Homocysteine, Methylmalonic Acid, and 2-Methylcitric Acid in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2010, 56, 1686–1695. [Google Scholar] [CrossRef] [Green Version]
- Naughten, E.R.; Yap, S.; Mayne, P.D. Newborn screening for homocystinuria: Irish and world experience. Eur. J. Nucl. Med. Mol. Imaging 1998, 157, S84–S87. [Google Scholar] [CrossRef] [PubMed]
- Sacharow, S.J.; Picker, J.D.; Levy, H.L. Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency. In Reviews in GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2004. [Google Scholar]
- Yap, S.; Naughten, E. Homocystinuria due to cystathionine β-synthase deficiency in Ireland: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J. Inherit. Metab. Dis. 1998, 21, 738–747. [Google Scholar] [CrossRef]
- Bowron, A.; Barton, A.; Scott, J.; Stansbie, D. Blood Spot Homocysteine: A Feasibility and Stability Study. Clin. Chem. 2005, 51, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.; Isaksson, A.; Hultberg, B. Homocysteine Export from Erythrocytes and Its Implication for Plasma Sampling. Clin. Chem. 1992, 38, 1311–1315. [Google Scholar] [CrossRef]
- Fiskerstrand, T.; Refsum, H.; Kvalheim, G.; Ueland, P.M. Homocysteine and other thiols in plasma and urine: Automated determination and sample stability. Clin. Chem. 1993, 39, 263–271. [Google Scholar] [CrossRef]
- Gramer, G.; Abdoh, G.; Ben-Omran, T.; Shahbeck, N.; Ali, R.; Mahmoud, L.; Fang-Hoffmann, J.; Hoffmann, G.F.; Al Rifai, H.; Okun, J.G. Newborn screening for remethylation disorders and vitamin B12 deficiency-evaluation of new strategies in cohorts from Qatar and Germany. World J. Pediatr. 2017, 13, 136–143. [Google Scholar] [CrossRef]
- Al-Dewik, N.; Ali, A.; Mahmoud, Y.; Shahbeck, N.; Ali, R.; Mahmoud, L.; Al-Mureikhi, M.; Al-Mesaifri, F.; Musa, S.; El-Akouri, K.; et al. Natural history, with clinical, biochemical, and molecular characterization of classical homocystinuria in the Qatari population. J. Inherit. Metab. Dis. 2019, 42, 818–830. [Google Scholar] [CrossRef]
- Yamada, K.; Yokoyama, K.; Aoki, K.; Taketani, T.; Yamaguchi, S. Long-Term Outcomes of Adult Patients with Homocystinuria before and after Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Moammar, H.; Cheriyan, G.; Mathew, R.; Al-Sannaa, N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann. Saudi Med. 2010, 30, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Arrayed, S.; Hamamy, H. The changing profile of consanguinity rates in bahrain, 1990–2009. J. Biosoc. Sci. 2011, 44, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Golbahar, J.; Al-Jishi, E.; Altayab, D.; Carreon, E.; Bakhiet, M.; Alkhayyat, H. Selective newborn screening of inborn errors of amino acids, organic acids and fatty acids metabolism in the Kingdom of Bahrain. Mol. Genet. Metab. 2013, 110, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Wasim, M.; Awan, F.R.; Khan, H.N.; Tawab, A.; Iqbal, M.; Ayesha, H. Aminoacidopathies: Prevalence, Etiology, Screening, and Treatment Options. Biochem. Genet. 2018, 56, 7–21. [Google Scholar] [CrossRef]
- Al-Hammadi, M.I. Presentation of Qatari Identity at National Museum of Qatar: Between Imagination and Reality. J. Conserv. Mus. Stud. 2018, 16. [Google Scholar] [CrossRef]
- Shibata, N.; Hasegawa, Y.; Yamada, K.; Kobayashi, H.; Purevsuren, J.; Yang, Y.; Dung, V.C.; Khanh, N.N.; Verma, I.C.; Bijarnia-Mahay, S.; et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective screening vs. expanded newborn screening. Mol. Genet. Metab. Rep. 2018, 16, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Faiyaz-Ul-Haque, M.; Shuaib, T.; Balobaid, A.; Rahbeeni, Z.; Abalkhail, H.; Al-Abdullatif, A.; Al-Hassnan, Z.; Peltekova, I.; Al-Owain, M. Clinical and molecular findings of 13 families from Saudi Arabia and a family from Sudan with homocystinuria. Clin. Genet. 2011, 81, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Silao, C.L.T.; Fabella, T.D.F.; Rama, K.I.D.; Estrada, S.C. Novel cystathionine β-synthase gene mutations in a Filipino patient with classic homocystinuria. Pediatr. Int. 2015, 57, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Attri, S.V.; Saini, A.G.; Sankhyan, N.; Singh, S.; Faruq, M.; Ramprasad, V.L.; Sharda, S.; Murugan, S. Seven novel genetic variants in a North Indian cohort with classical homocystinuria. Sci. Rep. 2020, 10, 17299. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Group | Disorder |
|---|---|
| Endocrinopathies | Congenital hypothyroidism |
| Congenital adrenal hyperplasia | |
| Aminoacidopathies | Argininosuccinic aciduria (ASA lyase deficiency) |
| Citrullinemia (ASA synthetase deficiency) | |
| Homocystinuria (cystathionine synthase def.) | |
| Maple syrup urine disease (MSUD) | |
| Phenylketonuria (PKU) | |
| Tyrosinemia (Type I) | |
| Fatty Acid Oxidation Disorders | Long chain hydroxy acyl-CoA dehydrogenase deficiency (LCHAD) |
| Medium chain acyl-CoA dehydrogenase def. (MCAD) | |
| Trifunctional protein deficiency (TFP) | |
| Very long chain acyl-CoA dehydrogenase deficiency (VLCAD) | |
| Organic Acidemias | 3-Methylcrotonyl-CoA carboxylase deficiency (3MCC) |
| 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency (3HMG-CoA lyase deficiency) | |
| Beta ketothiolase deficiency (mitochondrial acetoacetyl CoA thiolase deficiency) | |
| Glutaric acidemia type I (GA-I)) | |
| Isovaleric acidemia (IVA) | |
| Methyl malonic acidemia (MMA) | |
| Multiple CoA carboxylase deficiency (MCD) | |
| Propionic acidemia (PA) | |
| Galactosemia | Classic galactosemia |
| Biotinidase Deficiency | |
| Hearing Loss | |
| Pulse Oximetry for CHD |
| 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | Total | |
|---|---|---|---|---|---|---|---|
| Total samples received in NBS laboratory | 56,441 | 56,333 | 55,210 | 59,655 | 57,951 | 52,789 | 338,379 |
| Total newborns screened in Kuwait | 51,315 | 50,916 | 48,501 | 53,689 | 52,155 | 47,510 | 304,086 |
| Total premature newborns screened ≤ 33 wks | 2823 | 3263 | 3312 | 3350 | 3495 | 3298 | 19,541 |
| Newborns ≤ 33 wks (with exclusive high methionine) | 35 | 28 | 43 | 46 | 16 | 12 | 180 |
| Newborns >33 wks (with exclusive high methionine) | 49 | 17 | 38 | 73 | 19 | 24 | 220 |
| No. of all newborns in Kuwait per CSB | NA | 53,565 | 56,121 | 59,172 | 58,797 | 59,271 | NA |
| No. of Kuwaiti newborns per CSB | NA | 32,263 | 33,168 | 33,680 | 33,431 | 33,581 | NA |
| No. of Non-Kuwaiti newborns per CSB | NA | 21,302 | 22,953 | 25,492 | 25,366 | 25,690 | NA |
| Screened Kuwaiti newborns | 29,762 * | 30,145 | 28,645 | 29,074 | 28,733 | 24,859 | 171,218 |
| Screened non-Kuwaiti newborns | 21,553 * | 20,771 | 19,856 | 24,615 | 23,422 | 22,651 | 132,868 |
| Newborns not screened under national NBS program | 0 ψ | 2649 | 7620 | 5483 | 6642 | 11,761 | 34,155 |
| Percent of coverage of national NBS program (%) | 100 ψ | 94.8 | 84.3 | 89.8 | 87.3 | 75.2 | 88.8 |
| Nationality | Sex | Date of Birth (Month. Year) | Gestational Age (Weeks) | Age at NBS Result (Days) | Age at Start of Treatment (Days) | * DNA Variant | Protein Variant | Zygosity | 1st DBS Met Levels (Cutoff 44 μmol/L) | Met/Phe (Cutoff 0.75) | tHcy Levels (Cutoff 15 μmol/L) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | KSA | M | 07.2015 | 39 | 5 | NA | NA | NA | NA | 52 | NA | NA |
| P2 | K | M | 01.2017 | 38 | 4 | 16 | c.969G>A | Trp323Ter | Homo | 94.2 | 1.5 | 98 |
| P3 | K | F | 05.2017 | 39 | 3 | 9 | c.969G>A | Trp323Ter | Homo | 75 | 1.37 | 147 |
| P4 | E | F | 03.2019 | 37 | 4 | 5 | c.982G>A | Asp328Asn | Homo | 119 | 1.59 | 132 |
| P5 | K | M | 05.2019 | 40 | 5 | 12 | c.1006C>T | Arg336Cys | Homo | 63.2 | 1.02 | 113 |
| P6 | K | M | 01.2020 | 37 | 4 | 7 | c.969G>A | Trp323Ter | Homo | 93.28 | 1.9 | 111 |
| Cutoff | Sensitivity | Specificity | No. (%) of Positives | PPV % |
|---|---|---|---|---|
| Met > 44 μmol/L | 1 | 0.998 | 512 э (0.15%) | 1.17 |
| Met > 44 μmol/L& Met/Phe > 0.75 | 1 | 0.999 | 174 (0.05%) | 3.4 |
| Met/Phe > 0.65 | 1 | 0.996 | 1190 (0.35%) | 0.5 |
| Met/Phe > 0.70 | 1 | 0.998 | 658 (0.19) | 0.9 |
| Met/Phe > 0.75 ψ | 1 | 0.998 | 417 (0.12%) | 1.4 |
| Met/Phe > 0.80 | 1 | 0.999 | 286 (0.08%) | 2.1 |
| Met/Phe > 0.85 | 1 | 0.999 | 206 (0.06%) | 2.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsharhan, H.; Ahmed, A.A.; Ali, N.M.; Alahmad, A.; Albash, B.; Elshafie, R.M.; Alkanderi, S.; Elkazzaz, U.M.; Cyril, P.X.; Abdelrahman, R.M.; et al. Early Diagnosis of Classic Homocystinuria in Kuwait through Newborn Screening: A 6-Year Experience. Int. J. Neonatal Screen. 2021, 7, 56. https://doi.org/10.3390/ijns7030056
Alsharhan H, Ahmed AA, Ali NM, Alahmad A, Albash B, Elshafie RM, Alkanderi S, Elkazzaz UM, Cyril PX, Abdelrahman RM, et al. Early Diagnosis of Classic Homocystinuria in Kuwait through Newborn Screening: A 6-Year Experience. International Journal of Neonatal Screening. 2021; 7(3):56. https://doi.org/10.3390/ijns7030056
Chicago/Turabian StyleAlsharhan, Hind, Amir A. Ahmed, Naser M. Ali, Ahmad Alahmad, Buthaina Albash, Reem M. Elshafie, Sumaya Alkanderi, Usama M. Elkazzaz, Parakkal Xavier Cyril, Rehab M. Abdelrahman, and et al. 2021. "Early Diagnosis of Classic Homocystinuria in Kuwait through Newborn Screening: A 6-Year Experience" International Journal of Neonatal Screening 7, no. 3: 56. https://doi.org/10.3390/ijns7030056
APA StyleAlsharhan, H., Ahmed, A. A., Ali, N. M., Alahmad, A., Albash, B., Elshafie, R. M., Alkanderi, S., Elkazzaz, U. M., Cyril, P. X., Abdelrahman, R. M., Elmonairy, A. A., Ibrahim, S. M., Elfeky, Y. M. E., Sadik, D. I., Al-Enezi, S. D., Salloum, A. M., Girish, Y., Al-Ali, M., Ramadan, D. G., ... Bastaki, L. (2021). Early Diagnosis of Classic Homocystinuria in Kuwait through Newborn Screening: A 6-Year Experience. International Journal of Neonatal Screening, 7(3), 56. https://doi.org/10.3390/ijns7030056