
Thirty-Year Lessons from the Newborn Screening for Congenital Adrenal Hyperplasia (CAH) in Japan


Abstract
1. Introduction
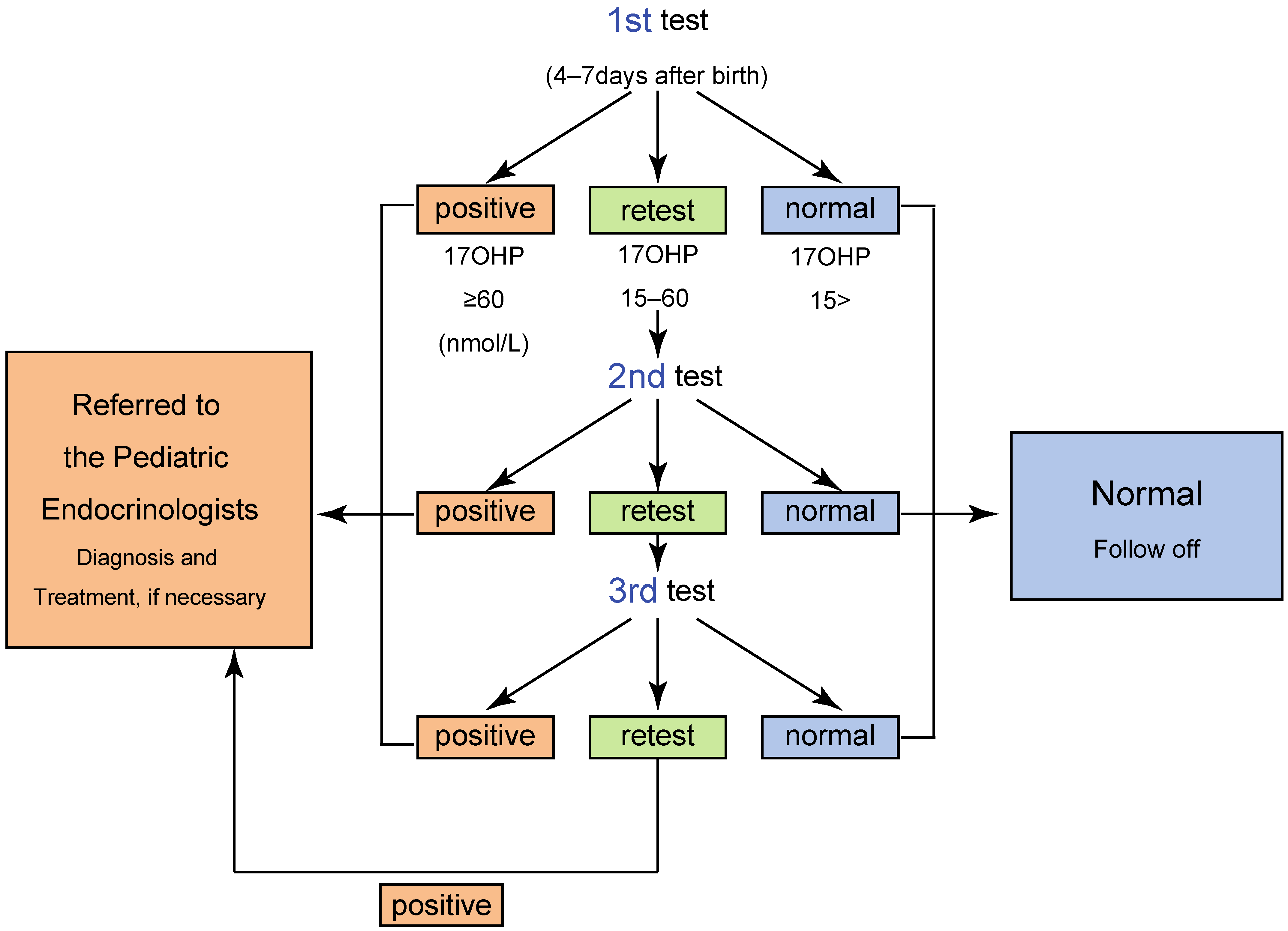
2. Screening System in Japan
3. Clinical Outcomes of the Newborn Screening for CAH in Japan
3.1. The Effects of the Screening
3.2. The Progression of Salt Wasting during the First Two Weeks of Life
3.3. Triage of the Neonates with Salt Wasting by Body Weight Change
4. Potential Issues of Testing Practices in the Newborn Screening for CAH in Japan
4.1. The Timeline of the Newborn Screening for 21OHD
4.2. High Rate of False Positive
4.3. LC-MS/MS Analysis of 17αOHP as a Second-Tier Test and Diagnostic Test for 21OHD
5. Database Composition and Improvement of Screening Program
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 17αOHP | 17-Hydroxyprogesterone |
| yrs | Years |
| CAH | Congenital adrenal hyperplasia |
| CH | Congenital hypothyroidism |
| PKU | Phenylketonuria |
| GAL | Galactosemia |
| AAD | Disorders of amino acid metabolism |
| OA | Disorders of organic acid metabolism |
| FAOD | Disorders of fatty acid metabolism |
References
- Speiser, P.W.; White, P.C. Congenital adrenal hyperplasia. N. Engl. J. Med. 2003, 349, 776–788. [Google Scholar] [CrossRef]
- White, P.C.; Speiser, P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 2000, 21, 245–291. [Google Scholar] [CrossRef] [PubMed]
- van der Kamp, H.J.; Wit, J.M. Neonatal screening for congenital adrenal hyperplasia. Eur. J. Endocrinol. 2004, 151 (Suppl. S3), U71–U75. [Google Scholar] [CrossRef]
- Suwa, S. Nationwide survey of neonatal mass-screening for congenital adrenal hyperplasia in Japan. Screening 1994, 3, 141–151. [Google Scholar] [CrossRef]
- Tsuji, A.; Konishi, K.; Hasegawa, S.; Anazawa, A.; Onishi, T.; Ono, M.; Morio, T.; Kitagawa, T.; Kashimada, K. Newborn screening for congenital adrenal hyperplasia in Tokyo, Japan from 1989 to 2013: A retrospective population-based study. BMC Pediatr. 2015, 15, 209. [Google Scholar] [CrossRef]
- Morikawa, S.; Nakamura, A.; Fujikura, K.; Fukushi, M.; Hotsubo, T.; Miyata, J.; Ishizu, K.; Tajima, T. Results from 28 years of newborn screening for congenital adrenal hyperplasia in sapporo. Clin. Pediatr. Endocrinol. 2014, 23, 35–43. [Google Scholar] [CrossRef][Green Version]
- Clayton, P.E.; Miller, W.L.; Oberfield, S.E.; Ritzen, E.M.; Sippell, W.G.; Speiser, P.W.; Group, E.L.C.W. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm. Res. 2002, 58, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Fujieda, K.; Nakae, J.; Mikami, A.; Cutler, G.B., Jr. Mutations of the CYP21 gene in nonclassical steroid 21-hydroxylase deficiency in Japan. Endocr. J. 1998, 45, 493–497. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tajima, T.; Fujieda, K.; Nakae, J.; Toyoura, T.; Shimozawa, K.; Kusuda, S.; Goji, K.; Nagashima, T.; Cutler, G.B., Jr. Molecular basis of nonclassical steroid 21-hydroxylase deficiency detected by neonatal mass screening in Japan. J. Clin. Endocrinol. Metab. 1997, 82, 2350–2356. [Google Scholar] [CrossRef]
- Kashimada, K.; Ishii, T.; Nagasaki, K.; Ono, M.; Tajima, T.; Yokota, I.; Hasegawa, Y. Clinical, biochemical, and genetic features of non-classical 21-hydroxylase deficiency in Japanese children. Endocr. J. 2015, 62, 277–282. [Google Scholar] [CrossRef]
- Tajima, T.; Fujikura, K.; Fukushi, M.; Hotsubo, T.; Mitsuhashi, Y. Neonatal screening for congenital adrenal hyperplasia in Japan. Pediatr. Endocrinol. Rev. 2012, 10 (Suppl. S1), 72–78. [Google Scholar] [PubMed]
- Tajima, T.; Fukushi, M. Neonatal mass screening for 21-hydroxylase deficiency. Clin. Pediatr. Endocrinol. 2016, 25, 1–8. [Google Scholar] [CrossRef]
- Gau, M.; Konishi, K.; Takasawa, K.; Nakagawa, R.; Tsuji-Hosokawa, A.; Hashimoto, A.; Sutani, A.; Tajima, T.; Hasegawa, T.; Morio, T.; et al. The progression of salt wasting and the body weight change during the first two weeks of life in classical 21-hydroxylase deficiency patients. Clin. Endocrinol. 2020, 94, 229–236. [Google Scholar] [CrossRef]
- Donaldson, M.D.; Thomas, P.H.; Love, J.G.; Murray, G.D.; McNinch, A.W.; Savage, D.C. Presentation, acute illness, and learning difficulties in salt wasting 21-hydroxylase deficiency. Arch. Dis. Child. 1994, 70, 214–218. [Google Scholar] [CrossRef][Green Version]
- Suwa, S. Congenital adrenal hyperplasia. Shouni Naika 1994, 26, 1967–1972. (In Japanese) [Google Scholar]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Suwa, S.; Maesaka, H.; Suzuki, J.; Katsumata, N. A suvery report: The comrbidities of congenital adrenal hyperplasia. In A Study for Newborn Screening, Annual Report in 1995; The Japanese Ministry of Health, Labour and Welfare’s Study Group for Physical and Mental Disabilities: Tokyo, Japan, 1985; pp. 274–277. (In Japanese) [Google Scholar]
- Heather, N.L.; Seneviratne, S.N.; Webster, D.; Derraik, J.G.; Jefferies, C.; Carll, J.; Jiang, Y.; Cutfield, W.S.; Hofman, P.L. Newborn screening for congenital adrenal hyperplasia in New Zealand, 1994–2013. J. Clin. Endocrinol. Metab. 2015, 100, 1002–1008. [Google Scholar] [CrossRef]
- Shima, R.; Sawano, K.; Shibata, N.; Nyuzuki, H.; Sasaki, S.; Sato, H.; Ogawa, Y.; Abe, Y.; Nagasaki, K.; Saitoh, A. Timing of hyponatremia development in patients with salt-wasting-type 21-hydroxylase deficiency. Clin. Pediatr. Endocrinol. 2020, 29, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Freedenberg, D.; Berry, S.; Dimmock, D.; Gibson, J.; Greene, C.; Kronn, D.; Tanksley, S. Society for Inherited Metabolic Disorders (SIMD) Position Statement, 2014: Identifying Abnormal Newborn Screens Requiring Immediate Notification of the Health Care Provider. Available online: https://www.simd.org/Issues/SIMD%20NBS%20Critical%20Conditions%20policy%20statement.pdf (accessed on 23 April 2021).
- HRSA, F.A.C. Newborn Screening Timeliness Goals. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/newborn-screening-timeliness.html (accessed on 20 May 2021).
- Sontag, M.K.; Miller, J.I.; McKasson, S.; Sheller, R.; Edelman, S.; Yusuf, C.; Singh, S.; Sarkar, D.; Bocchini, J.; Scott, J.; et al. Newborn screening timeliness quality improvement initiative: Impact of national recommendations and data repository. PLoS ONE 2020, 15, e0231050. [Google Scholar] [CrossRef] [PubMed]
- Loeber, J.G. Current Status of Newborn Screening in Europe (Based on an ISNS Survey Held during 2018). Available online: https://www.isns-neoscreening.org/wp-content/uploads/2019/04/NBS-in-Europe-2018.pdf (accessed on 5 October 2020).
- Held, P.K.; Bird, I.M.; Heather, N.L. Newborn screening for congenital adrenal hyperplasia: Review of factors affecting screening accuracy. Int. J. Neonatal. Screen 2020, 6, 67. [Google Scholar] [CrossRef]
- Loeber, J.G.; Platis, D.; Zetterstrom, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal screening in Europe revisited: An ISNS perspective on the current state and developments since 2010. Int. J. Neonatal. Screen 2021, 7, 15. [Google Scholar] [CrossRef]
- Coulm, B.; Coste, J.; Tardy, V.; Ecosse, E.; Roussey, M.; Morel, Y.; Carel, J.C.; Group, D.S. Efficiency of neonatal screening for congenital adrenal hyperplasia due to 21-hydroxylase deficiency in children born in mainland France between 1996 and 2003. Arch. Pediatr. Adolesc. Med. 2012, 166, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Pode-Shakked, N.; Blau, A.; Pode-Shakked, B.; Tiosano, D.; Weintrob, N.; Eyal, O.; Zung, A.; Levy-Khademi, F.; Tenenbaum-Rakover, Y.; Zangen, D.; et al. Combined gestational age- and birth weight-adjusted cutoffs for newborn screening of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2019, 104, 3172–3180. [Google Scholar] [CrossRef]
- van der Kamp, H.J.; Oudshoorn, C.G.; Elvers, B.H.; van Baarle, M.; Otten, B.J.; Wit, J.M.; Verkerk, P.H. Cutoff levels of 17-alpha-hydroxyprogesterone in neonatal screening for congenital adrenal hyperplasia should be based on gestational age rather than on birth weight. J. Clin. Endocrinol. Metab. 2005, 90, 3904–3907. [Google Scholar] [CrossRef]
- Nordenstrom, A.; Wedell, A.; Hagenfeldt, L.; Marcus, C.; Larsson, A. Neonatal screening for congenital adrenal hyperplasia: 17-hydroxyprogesterone levels and CYP21 genotypes in preterm infants. Pediatrics 2001, 108, e68. [Google Scholar] [CrossRef]
- Allen, D.B.; Hoffman, G.L.; Fitzpatrick, P.; Laessig, R.; Maby, S.; Slyper, A. Improved precision of newborn screening for congenital adrenal hyperplasia using weight-adjusted criteria for 17-hydroxyprogesterone levels. J. Pediatr. 1997, 130, 128–133. [Google Scholar] [CrossRef]
- Miller, W.L. Congenital adrenal hyperplasia: Time to replace 17OHP with 21-deoxycortisol. Horm. Res. Paediatr. 2019, 91, 416–420. [Google Scholar] [CrossRef]
- Takasawa, K.; Ono, M.; Hijikata, A.; Matsubara, Y.; Katsumata, N.; Takagi, M.; Morio, T.; Ohara, O.; Kashimada, K.; Mizutani, S. Two novel HSD3B2 missense mutations with diverse residual enzymatic activities for Delta5-steroids. Clin. Endocrinol. 2014, 80, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Nishio, T.; Uchida, S.; Shimada, K.; Fukushi, M.; Maeda, M. Simultaneous determination of 17alpha-hydroxypregnenolone and 17alpha-hydroxyprogesterone in dried blood spots from low birth weight infants using LC-MS/MS. J. Pharm. Biomed. Anal. 2008, 48, 177–182. [Google Scholar] [CrossRef]
- Janzen, N.; Peter, M.; Sander, S.; Steuerwald, U.; Terhardt, M.; Holtkamp, U.; Sander, J. Newborn screening for congenital adrenal hyperplasia: Additional steroid profile using liquid chromatography-tandem mass spectrometry. J. Clin. Endocrinol. Metab. 2007, 92, 2581–2589. [Google Scholar] [CrossRef]
- Kulle, A.E.; Riepe, F.G.; Hedderich, J.; Sippell, W.G.; Schmitz, J.; Niermeyer, L.; Holterhus, P.M. LC-MS/MS based determination of basal- and ACTH-stimulated plasma concentrations of 11 steroid hormones: Implications for detecting heterozygote CYP21A2 mutation carriers. Eur. J. Endocrinol. 2015, 173, 517–524. [Google Scholar] [CrossRef]
- Monostori, P.; Szabo, P.; Marginean, O.; Bereczki, C.; Karg, E. Concurrent confirmation and differential diagnosis of congenital adrenal hyperplasia from dried blood spots: Application of a second-tier LC-MS/MS assay in a cross-border cooperation for newborn screening. Horm. Res. Paediatr. 2015, 84, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Tsai, C.H.; Tsai, F.J.; Wu, J.Y.; Lin, W.D.; Lee, C.C. Rapid screening assay of congenital adrenal hyperplasia by measuring 17 alpha-hydroxyprogesterone with high-performance liquid chromatography/electrospray ionization tandem mass spectrometry from dried blood spots. J. Clin. Lab. Anal. 2002, 16, 20–25. [Google Scholar] [CrossRef]
- Lacey, J.M.; Minutti, C.Z.; Magera, M.J.; Tauscher, A.L.; Casetta, B.; McCann, M.; Lymp, J.; Hahn, S.H.; Rinaldo, P.; Matern, D. Improved specificity of newborn screening for congenital adrenal hyperplasia by second-tier steroid profiling using tandem mass spectrometry. Clin. Chem. 2004, 50, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.Z.; Lacey, J.M.; Magera, M.J.; Hahn, S.H.; McCann, M.; Schulze, A.; Cheillan, D.; Dorche, C.; Chace, D.H.; Lymp, J.F.; et al. Steroid profiling by tandem mass spectrometry improves the positive predictive value of newborn screening for congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2004, 89, 3687–3693. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, M.; Isobe, M.; Kanda, K.; Saraie, H.; Fujikura, K.; Mitsui, N.; Mae, H.; Yamagami, Y. The quality control of LC-MS/MS screening for CAH as the second tier test. In Proceedings of the 47th Annual meeting of Japan Society for Neonatal Screening, Gifu, Japan, 25–26 September 2020. [Google Scholar]
- Children and Families Bureau, Labour and Welfare in Japan. Notification for Newborn Screening, 2018. Notification from Director of the Maternal and Child Health Division. Available online: https://www.jsms.gr.jp/download/MHLW_MCH_20180330.pdf (accessed on 23 April 2021). (In Japanese)
- Therrell, B.L., Jr.; Berenbaum, S.A.; Manter-Kapanke, V.; Simmank, J.; Korman, K.; Prentice, L.; Gonzalez, J.; Gunn, S. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics 1998, 101, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Honour, J.W.; Anderson, J.M.; Shackleton, C.H. Difficulties in the diagnosis of congenital adrenal hyperplasia in early infancy: The 11 beta-hydroxylase defect. Acta Endocrinol. 1983, 103, 101–109. [Google Scholar] [CrossRef]
- Tonetto-Fernandes, V.; Lemos-Marini, S.H.; Kuperman, H.; Ribeiro-Neto, L.M.; Verreschi, I.T.; Kater, C.E. Serum 21-Deoxycortisol, 17-Hydroxyprogesterone, and 11-deoxycortisol in classic congenital adrenal hyperplasia: Clinical and hormonal correlations and identification of patients with 11beta-hydroxylase deficiency among a large group with alleged 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2006, 91, 2179–2184. [Google Scholar] [CrossRef][Green Version]
- Homma, K.; Hasegawa, T.; Nagai, T.; Adachi, M.; Horikawa, R.; Fujiwara, I.; Tajima, T.; Takeda, R.; Fukami, M.; Ogata, T. Urine steroid hormone profile analysis in cytochrome P450 oxidoreductase deficiency: Implication for the backdoor pathway to dihydrotestosterone. J. Clin. Endocrinol. Metab. 2006, 91, 2643–2649. [Google Scholar] [CrossRef]
- Fluck, C.E.; Tajima, T.; Pandey, A.V.; Arlt, W.; Okuhara, K.; Verge, C.F.; Jabs, E.W.; Mendonca, B.B.; Fujieda, K.; Miller, W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004, 36, 228–230. [Google Scholar] [CrossRef]
- Jeandron, D.D.; Sahakitrungruang, T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic 3beta-hydroxysteroid dehydrogenase deficiency: An unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm. Res. Paediatr. 2012, 77, 334–338. [Google Scholar] [CrossRef]
- Cara, J.F.; Moshang, T., Jr.; Bongiovanni, A.M.; Marx, B.S. Elevated 17-hydroxyprogesterone and testosterone in a newborn with 3-beta-hydroxysteroid dehydrogenase deficiency. N. Engl. J. Med. 1985, 313, 618–621. [Google Scholar] [CrossRef]
- Homma, K.; Hasegawa, T.; Takeshita, E.; Watanabe, K.; Anzo, M.; Toyoura, T.; Jinno, K.; Ohashi, T.; Hamajima, T.; Takahashi, Y.; et al. Elevated urine pregnanetriolone definitively establishes the diagnosis of classical 21-hydroxylase deficiency in term and preterm neonates. J. Clin. Endocrinol. Metab. 2004, 89, 6087–6091. [Google Scholar] [CrossRef]
- Pignatelli, D.; Carvalho, B.L.; Palmeiro, A.; Barros, A.; Guerreiro, S.G.; Macut, D. The Complexities in Genotyping of Congenital Adrenal Hyperplasia: 21-Hydroxylase Deficiency. Front. Endocrinol. 2019, 10, 432. [Google Scholar] [CrossRef] [PubMed]
- Parajes, S.; Quinterio, C.; Dominguez, F.; Loidi, L. A simple and robust quantitative PCR assay to determine CYP21A2 gene dose in the diagnosis of 21-hydroxylase deficiency. Clin. Chem. 2007, 53, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lu, L.; Yu, B.; Mao, J.; Wang, X.; Nie, M.; Wu, X. The prevalence of the chimeric TNXA/TNXB gene and clinical symptoms of ehlers-danlos syndrome with 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2020, 105, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Janzen, N.; Riepe, F.G.; Peter, M.; Sander, S.; Steuerwald, U.; Korsch, E.; Krull, F.; Muller, H.L.; Heger, S.; Brack, C.; et al. Neonatal screening: Identification of children with 11beta-hydroxylase deficiency by second-tier testing. Horm. Res. Paediatr. 2012, 77, 195–199. [Google Scholar] [CrossRef]
- Koyama, Y.; Homma, K.; Fukami, M.; Miwa, M.; Ikeda, K.; Ogata, T.; Hasegawa, T.; Murata, M. Two-step biochemical differential diagnosis of classic 21-hydroxylase deficiency and cytochrome P450 oxidoreductase deficiency in Japanese infants by GC-MS measurement of urinary pregnanetriolone/ tetrahydroxycortisone ratio and 11beta-hydroxyandrosterone. Clin. Chem. 2012, 58, 741–747. [Google Scholar] [CrossRef]
- Burgard, P.; Rupp, K.; Lindner, M.; Haege, G.; Rigter, T.; Weinreich, S.S.; Loeber, J.G.; Taruscio, D.; Vittozzi, L.; Cornel, M.C.; et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 2. From screening laboratory results to treatment, follow-up and quality assurance. J. Inherit. Metab. Dis. 2012, 35, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Advisory Committee on Heritable Disorders in Newborns and Children; Zuckerman, A.E.; Badawi, D.; Brosco, J.P.; Brower, A.; Eichwald, J.; Feuchtbaum, L.; Finitzo, T.; Flannery, D.; Green, N.; et al. The Role of Quality Measures to Promote Long-Term Follow-up of Children Identified by Newborn Screening Programs; Advisory Committee on Heritable Disorders in Newborns and Children Advisory Committee on Heritable Disorders in Newborns and Children. 2018. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/reports-recommendations/reports/role-of-quality-measures-in-nbs-sept2018-508c.pdf” (accessed on 23 April 2021).
- Stikkelbroeck, N.M.; Oyen, W.J.; van der Wilt, G.J.; Hermus, A.R.; Otten, B.J. Normal bone mineral density and lean body mass, but increased fat mass, in young adult patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2003, 88, 1036–1042. [Google Scholar] [CrossRef]
- Charmandari, E.; Chrousos, G.P. Metabolic syndrome manifestations in classic congenital adrenal hyperplasia: Do they predispose to atherosclerotic cardiovascular disease and secondary polycystic ovary syndrome? Ann. N. Y. Acad. Sci. 2006, 1083, 37–53. [Google Scholar] [CrossRef]
- Zimmermann, A.; Grigorescu-Sido, P.; AlKhzouz, C.; Patberg, K.; Bucerzan, S.; Schulze, E.; Zimmermann, T.; Rossmann, H.; Geiss, H.C.; Lackner, K.J.; et al. Alterations in lipid and carbohydrate metabolism in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm. Res. Paediatr. 2010, 74, 41–49. [Google Scholar] [CrossRef]
- Finkielstain, G.P.; Kim, M.S.; Sinaii, N.; Nishitani, M.; Van Ryzin, C.; Hill, S.C.; Reynolds, J.C.; Hanna, R.M.; Merke, D.P. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2012, 97, 4429–4438. [Google Scholar] [CrossRef] [PubMed]
- Merke, D.P.; Bornstein, S.R. Congenital adrenal hyperplasia. Lancet 2005, 365, 2125–2136. [Google Scholar] [CrossRef]
- Khalid, J.M.; Oerton, J.M.; Dezateux, C.; Hindmarsh, P.C.; Kelnar, C.J.; Knowles, R.L. Incidence and clinical features of congenital adrenal hyperplasia in Great Britain. Arch. Dis. Child. 2012, 97, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Takishima, S.; Nakajima, K.; Nomura, R.; Tsuji-Hosokawa, A.; Matsuda, N.; Matsubara, Y.; Ono, M.; Miyai, K.; Takasawa, K.; Morio, T.; et al. Lower body weight and BMI at birth were associated with early adiposity rebound in 21-hydroxylase deficiency patients. Endocr. J. 2016, 63, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Pasterski, V.; Zucker, K.J.; Hindmarsh, P.C.; Hughes, I.A.; Acerini, C.; Spencer, D.; Neufeld, S.; Hines, M. Increased cross-gender identification independent of gender role behavior in girls with congenital adrenal hyperplasia: Results from a standardized assessment of 4- to 11-year-old children. Arch. Sex. Behav. 2015, 44, 1363–1375. [Google Scholar] [CrossRef]
- Meyer-Bahlburg, H.F.; Dolezal, C.; Baker, S.W.; Ehrhardt, A.A.; New, M.I. Gender development in women with congenital adrenal hyperplasia as a function of disorder severity. Arch. Sex. Behav. 2006, 35, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Bahlburg, H.F.; Dolezal, C.; Baker, S.W.; New, M.I. Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Arch. Sex. Behav. 2008, 37, 85–99. [Google Scholar] [CrossRef]
- Harasymiw, L.A.; Grosse, S.D.; Sarafoglou, K. Attention-deficit/hyperactivity disorder among US children and adolescents with congenital adrenal hyperplasia. J. Endocr. Soc. 2020, 4, bvaa152. [Google Scholar] [CrossRef]
- Hamed, S.A.; Metwalley, K.A.; Farghaly, H.S. Cognitive function in children with classic congenital adrenal hyperplasia. Eur. J. Pediatr. 2018, 177, 1633–1640. [Google Scholar] [CrossRef]
- Fox, D.A.; Ronsley, R.; Khowaja, A.R.; Haim, A.; Vallance, H.; Sinclair, G.; Amed, S. Clinical impact and cost efficacy of newborn screening for congenital adrenal hyperplasia. J. Pediatr. 2020, 220, 101–108.e2. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.E.; Downs, S.M. Comprehensive cost-utility analysis of newborn screening strategies. Pediatrics 2006, 117, S287–S295. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.K.; Grosse, S.D. The cost effectiveness of screening newborns for congenital adrenal hyperplasia. Public Health Genom. 2009, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Huemer, M.; Diodato, D.; Martinelli, D.; Olivieri, G.; Blom, H.; Gleich, F.; Kolker, S.; Kozich, V.; Morris, A.A.; Seifert, B.; et al. Phenotype, treatment practice and outcome in the cobalamin-dependent remethylation disorders and MTHFR deficiency: Data from the E-HOD registry. J. Inherit. Metab. Dis. 2019, 42, 333–352. [Google Scholar] [CrossRef]
- Posset, R.; Garbade, S.F.; Gleich, F.; Gropman, A.L.; de Lonlay, P.; Hoffmann, G.F.; Garcia-Cazorla, A.; Nagamani, S.C.S.; Baumgartner, M.R.; Schulze, A.; et al. Long-term effects of medical management on growth and weight in individuals with urea cycle disorders. Sci. Rep. 2020, 10, 11948. [Google Scholar] [CrossRef]
- Kolker, S.; Dobbelaere, D.; Haberle, J.; Burgard, P.; Gleich, F.; Summar, M.L.; Hannigan, S.; Parker, S.; Chakrapani, A.; Baumgartner, M.R.; et al. Networking across borders for individuals with organic acidurias and urea cycle disorders: The E-IMD consortium. JIMD Rep. 2015, 22, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kozich, V.; Sokolova, J.; Morris, A.A.M.; Pavlikova, M.; Gleich, F.; Kolker, S.; Krijt, J.; Dionisi-Vici, C.; Baumgartner, M.R.; Blom, H.J.; et al. Cystathionine beta-synthase deficiency in the E-HOD registry-part I: Pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis. J. Inherit. Metab. Dis. 2021, 44, 677–692. [Google Scholar] [CrossRef]
- Hoff, T.; Hoyt, A.; Therrell, B.; Ayoob, M. Exploring barriers to long-term follow-up in newborn screening programs. Genet. Med. 2006, 8, 563–570. [Google Scholar] [CrossRef]
- Loeber, J.G.; Burgard, P.; Cornel, M.C.; Rigter, T.; Weinreich, S.S.; Rupp, K.; Hoffmann, G.F.; Vittozzi, L. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J. Inherit. Metab. Dis. 2012, 35, 603–611. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
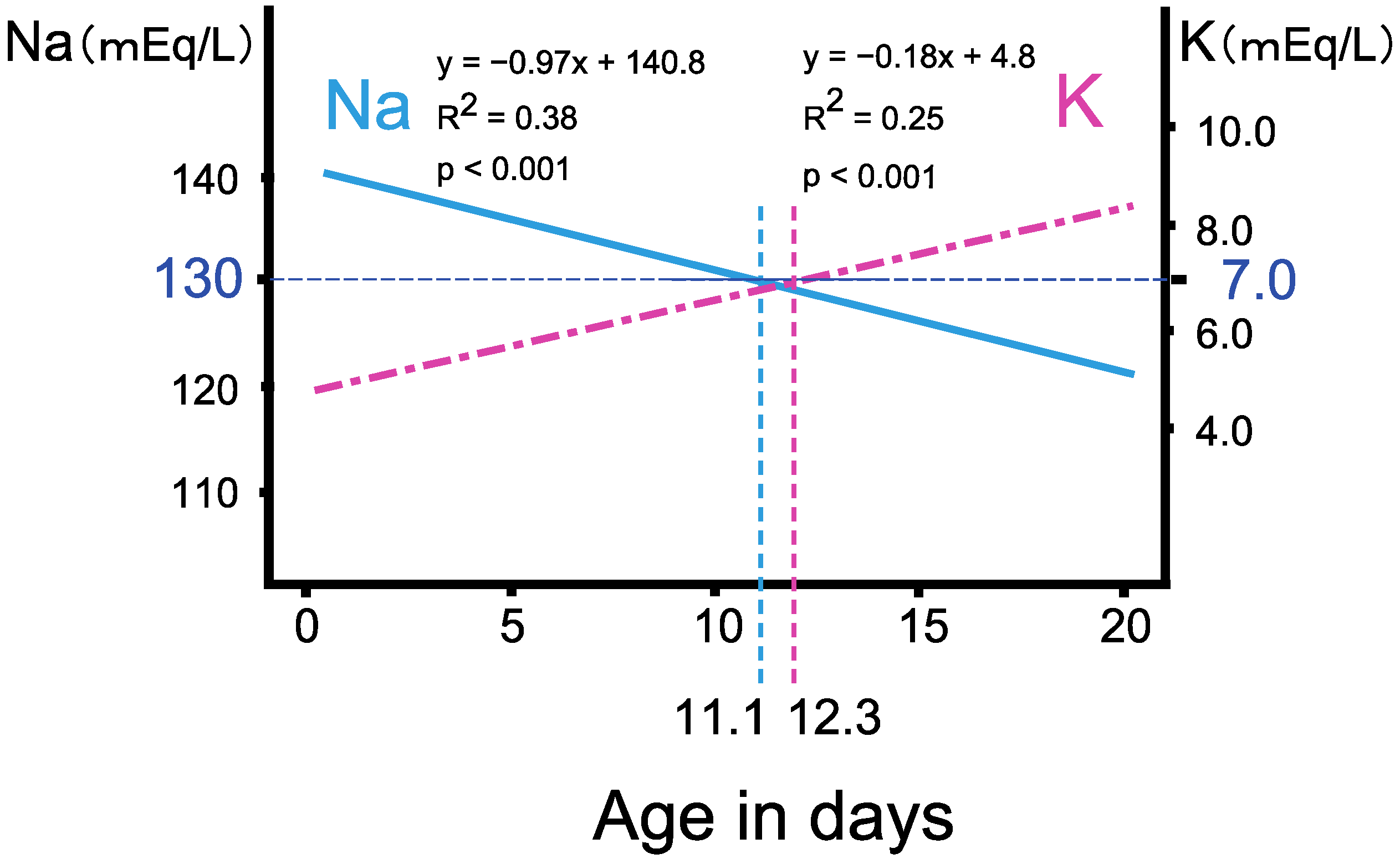{kind=link}
{kind=link}
{kind=link}
| <Criteria According to the Gestational Age> | |||||
|---|---|---|---|---|---|
| Gestational age at birth (weeks) *1a | ≤29 | 30–34 | 35–36 | ≥37 | |
| Corrected gestational age (weeks) *1b | ≤31 | 32–35 | 36–37 | ≥38 | |
| <Criteria According to Weight> *2,*3 | |||||
| Body weight (g) | ≤999 | 1000–1999 | 2000–2499 | ≥2500 | |
| Cutoff level of 17αOHP [n·mol/L] | Retest *4 | 60 | 45 | 24 | 15 |
| Positive *5 | 60 | 60 | 60 | ||
| Before CAH Screening * | After CAH Screening ** | |||||
|---|---|---|---|---|---|---|
| Male | Female | Total | Male | Female | Total | |
| SW type | 63 days (120) | 47 days (96) | 55 days (216) | 9.0 days (55) | 6.2 days (45) | 7.6 days (100) |
| SV type | 5.0 yrs (39) | 6.5 yrs (150) | 6.4 yrs (189) | |||
| Countries *1 | Approximate Population (Million) | Screening Panel | Interval Birth-Sampling | Interval Sampling Analysis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAH | CH | PKU | GAL | AAD, OA, FAOD | <48 h | 48–72 h | 72–96 h | >96 h | 1 d | 2 d | 3 d | 4 d | 5 d | >6 d | ||
| Austria | 8.8 | x *2 | x | x | x | >6 | x | x | x | x | x | |||||
| Belgium | 10.5 | x | x | x | x | >6 | x | x | x | x | ||||||
| Denmark | 5.6 | x | x | x | x | >6 | x | x | x | |||||||
| France | 67 | x | x | x | x | P *2 | x | x | x | |||||||
| Germany | 80 | x | x | x | x | >6 | x | x | x | x | ||||||
| Netherlands | 17.8 | x | x | x | x | >6 | x | x | x | x | ||||||
| Spain | 46.5 | x | x | x | P | >6 | x | x | x | x | x | x | x | |||
| Sweden | 10 | x | x | x | x | >6 | x | x | x | x | ||||||
| Switzerland | 8.1 | x | x | x | x | 1–6 | x | x | ||||||||
| Finland | 5.5 | x | x | >6 | x *4 | x | x | x | x | x | x | x | ||||
| Greece | 10.5 | x | x | x | x | x | ||||||||||
| Hungary | 10 | x | x | x | >6 | x | x | x | ||||||||
| Ireland | 4.9 | x | x | x | x | x | x | x | ||||||||
| Italy | 60.5 | x | x | P | P | x | x | x | x | x | ||||||
| Norway | 5.3 | x | x | >6 | x | x | ||||||||||
| Portugal | 10.3 | x | x | >6 | x | x | x | x | ||||||||
| U.K. | 66.6 | x | x | 1–6 | x | x | x | |||||||||
| U.S. *3 | 328.2 | 50/50 | 50/50 | 50/50 | 50/50 | 50/50 | x | x | x | |||||||
| JPN | 126.3 | x | x | x | x | >6 | x | x | x | x | ||||||
| Screening Positive Cutoff Level | |||||
|---|---|---|---|---|---|
| Prefecture | Saitama | Sapporo | Tokyo | ||
| Criteria A *2,*3 | Criteria B *2,*3 | ||||
| 17αOHP (ng/mL) | Term birth | >20 | >20 | >5 | >5 |
| Preterm birth | >30 | >50 | |||
| 21DOF (ng/mL) | >1.0 | >2.0 | >1.0 | ||
| (17αOHP + 4AD)/F | >2.0 | ||||
| 11DOF/17αOHP | <0.1 | ||||
| Retest Cutoff Level | |||||
| Prefecture | Saitama *2 | Sapporo *2 | Tokyo *2 | ||
| 17αOHP (ng/mL) | >1.0 | >2.5 | >1.5 | ||
| (17αOHP + 4AD)/F | >0.1 | >0.1 | >0.3 | ||
| 11DOF/17αOHP | <0.3 | <0.2 | <0.3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuji-Hosokawa, A.; Kashimada, K. Thirty-Year Lessons from the Newborn Screening for Congenital Adrenal Hyperplasia (CAH) in Japan. Int. J. Neonatal Screen. 2021, 7, 36. https://doi.org/10.3390/ijns7030036
Tsuji-Hosokawa A, Kashimada K. Thirty-Year Lessons from the Newborn Screening for Congenital Adrenal Hyperplasia (CAH) in Japan. International Journal of Neonatal Screening. 2021; 7(3):36. https://doi.org/10.3390/ijns7030036
Chicago/Turabian StyleTsuji-Hosokawa, Atsumi, and Kenichi Kashimada. 2021. "Thirty-Year Lessons from the Newborn Screening for Congenital Adrenal Hyperplasia (CAH) in Japan" International Journal of Neonatal Screening 7, no. 3: 36. https://doi.org/10.3390/ijns7030036
APA StyleTsuji-Hosokawa, A., & Kashimada, K. (2021). Thirty-Year Lessons from the Newborn Screening for Congenital Adrenal Hyperplasia (CAH) in Japan. International Journal of Neonatal Screening, 7(3), 36. https://doi.org/10.3390/ijns7030036